What is Apoptosis
Apoptosis is a form of programmed cell death that occurs in multicellular organisms and has multiple functions in ontogenetic development and tissue remodelling.
The process leads to characteristic cellular changes (morphology) and eventual death. The hallmarks for apoptosis include blebbing, cytoplasm shrinkage, nuclear fragmentation, chromatin condensation, chromosomal DNA fragmentation, and mRNA degeneration.
Apoptosis can also be visualised by the production of membrane-bound apoptotic bodies and an increase in the concentration of specific cleaved or degraded cellular proteins.
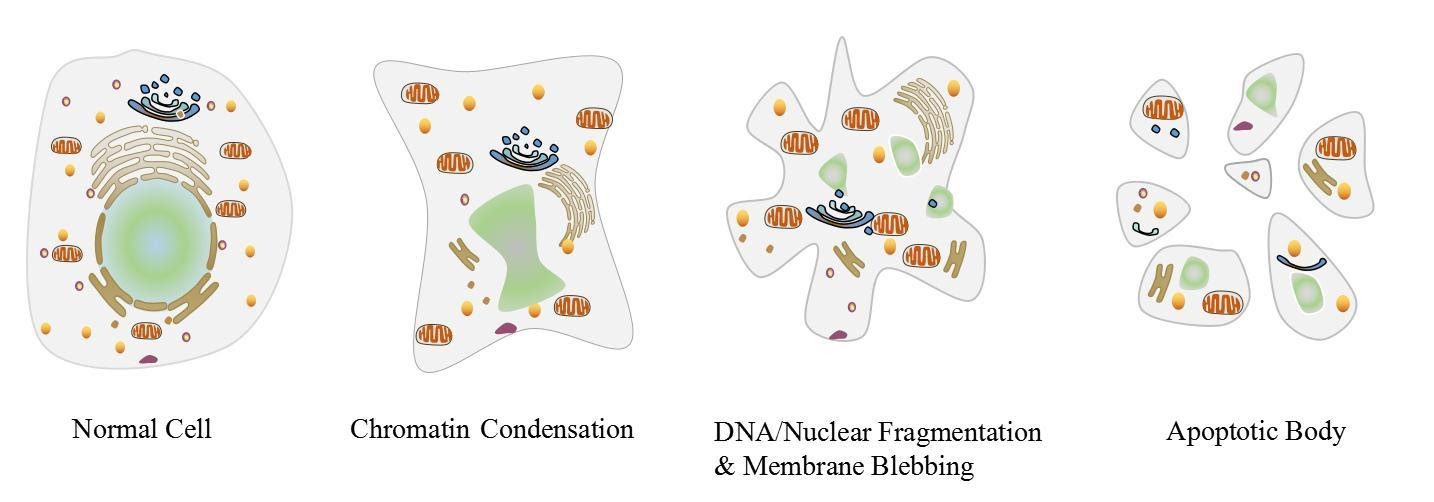
Figure 1: Visual representation of a cell undergoing apoptosis
All of the above characteristics can be measured or observed using various assays and kits to identify and quantify cell death within samples. The molecular players involved in this pathway are extensive (as shown in figure 2) but for this review, we will focus on the important protein families that are heavily regarded to either initiate or inhibit apoptosis.
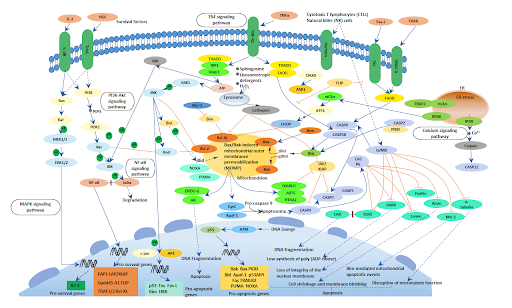
Figure 2: Overview of various apoptotic pathways
Caspases & the Bcl-2 family
Two major protein families regulate apoptotic pathways; the caspase family and the Bcl-2 family. Once activated, these molecules determine the fate of the cell. Caspase proteins are a group of cysteine-aspartic proteases that either falls into the subcategories of pro inflammatory caspases (caspase -1, -4, 5 and 12), initiator caspases (caspase -2, -8, -9 and -10) and executioner caspases (caspase -3, -6 and -7).
The caspase family thus provides critical links in cell regulatory networks and are integral components in supervising inflammation and cell death. The activation of caspase proteins are tightly controlled by the inactivation or activation of substrates and signalling events promoting their aggregation into dimers or macromolecular complexes, leading to their catalytic activation and the downstream demolition of cellular components and finally, apoptosis.
The Bcl-2 proteins (B-cell lymphoma 2) are believed to negotiate whether the cell commits to apoptosis or aborts the process. It is thought that the main mechanism of action of the Bcl-2 family is the regulation of cytochrome c release from the mitochondria via alteration of mitochondrial membrane permeability.
The Bcl-2 family therefore govern mitochondrial membrane permeability and can be either pro-apoptotic or antiapoptotic.
Bcl-2 proteins thus regulate the permeabilisation of mitochondria, which leads to the activation of the downstream caspase cascade events. The Bcl-2 family is subdivided into pro-apoptotic proteins (e.g. Bak, Bax, Bid, Bim, Noxa, PUMA) and anti apoptotic proteins (e.g. Bcl-2, Mcl-1, Bcl-XL, etc).
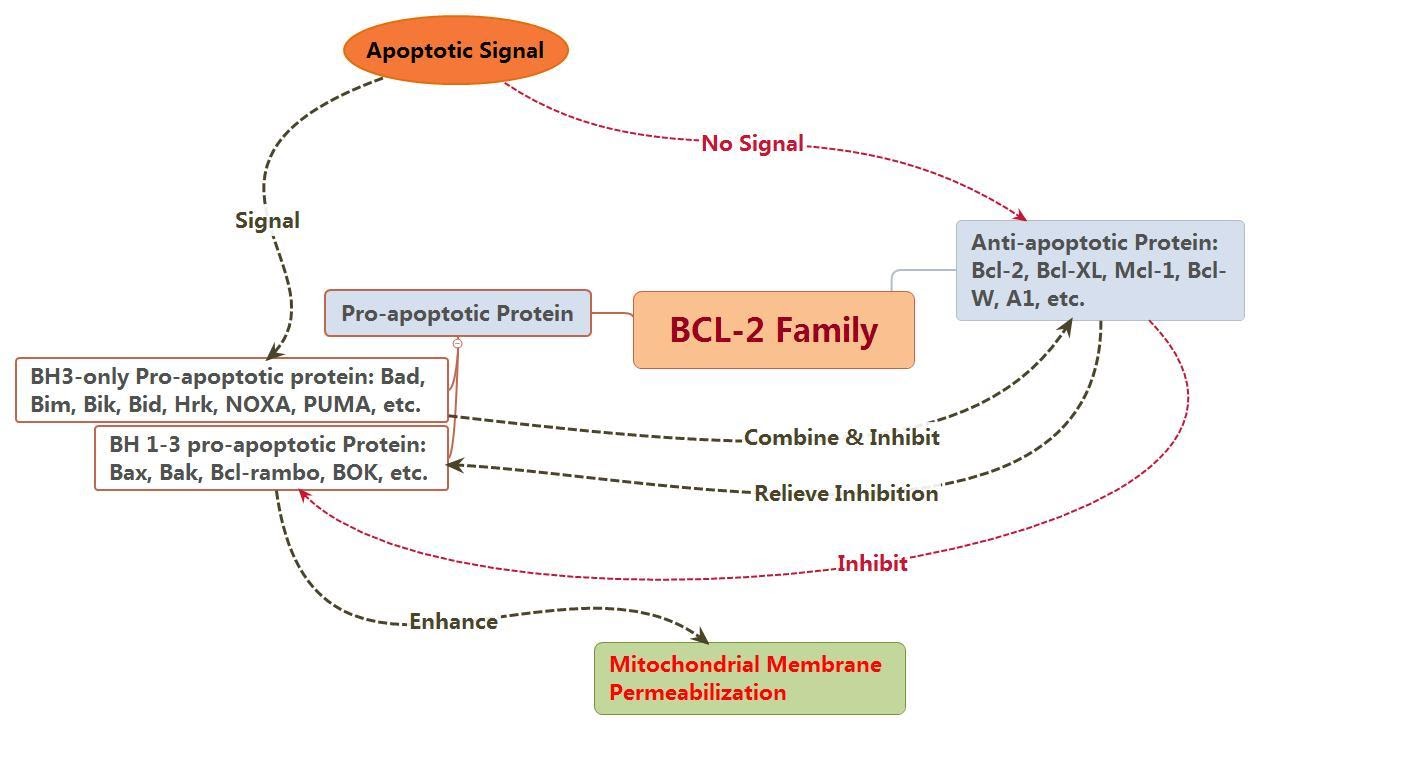 Figure 3: Depicting the pro & anti – apoptotic influences of the BCL-2 family members
Figure 3: Depicting the pro & anti – apoptotic influences of the BCL-2 family members
The Intrinsic & Extrinsic Pathway
One of the major triggers to stimulate apoptosis is the ‘Intrinsic pathway’ and the ‘Extrinsic pathway’. While the major catalyst for the intrinsic pathway is mitochondrial damage and subsequent release of pro-apoptotic proteins, the extrinsic pathway is capable directly activating effector caspases but can also cross talk with the intrinsic pathway leading to mitochondrial disruption, ultimately leading to the formation of the apoptosome and activation of caspases, leading to apoptosis.
In apoptosis initiated by death receptor signalling, the extrinsic pathway, mitochondrial damage is not required in most cells as caspase 8 can directly activate downstream caspases. The cell surface death receptors include Fas (CD95/APO1), TRAIL (TNF related apoptosis inducing ligand), death receptor 5 (DR5; also known as TNFRSF10B) receptors and TNFα (tumor necrosis factor-α) receptors.
These receptors can be activated by their respective ligands, such as CD95 ligand, TNFα as well as TRAIL. Once activated, corresponding receptors begin to activate and the recruitment of adaptor proteins such as adaptor FADD (FAS-associated DEATH domain protein), or in the case of TNFR1 the adaptor TRADD (TNFR1-associated DEATH domain protein), begin to complex with inactive caspase 8, leading to the formation of DISC (death-inducing signaling complex).
DISC can activate caspase 8, this active caspase 8 is then capable of cleaving BID to tBID, thus leading to the downstream activation of BAX/BAK, and the intrinsic apoptosis pathway, although it can also activate caspase 3 and 7 independently, thus bypassing the intrinsic pathway.
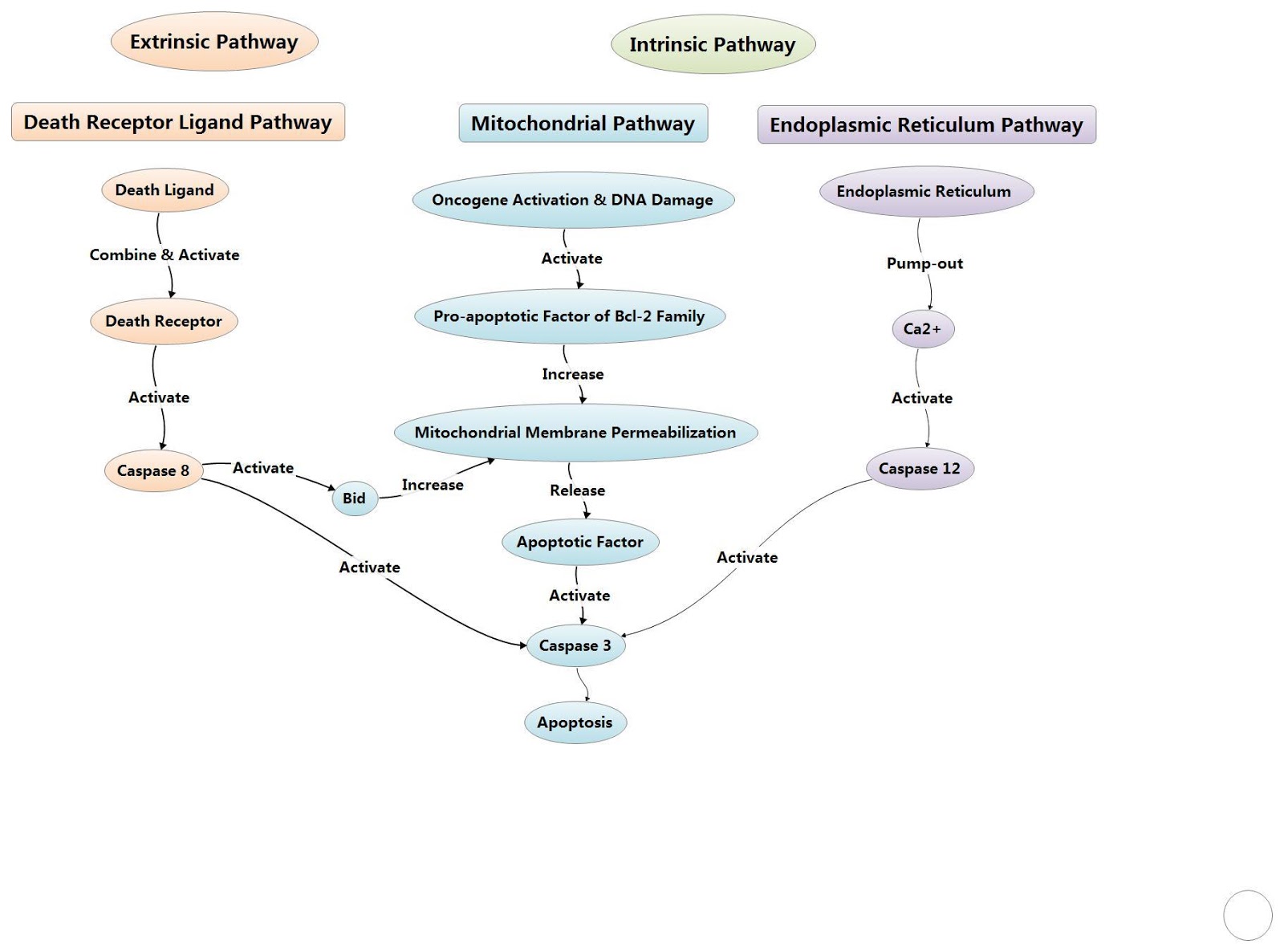
Figure 4: Overview of the extrinsic and intrinsic pathway
Mitochondrial Stress
External stimuli have been shown to compromise the permeability of the mitochondrial membrane, although the exact mechanisms have not been fully realised. Caspase 2 has been shown to become active after exposure to negative stimuli and once active can cleave and activate the pro-apoptotic protein Bid.
The active Bid subsequently compromises mitochondrial permeability, leading to the release of a variety of proteins including cytochrome c into the cytosol, which disturbs the homeostasis of additional pro- and anti-apoptotic, Bcl-2 proteins. Cytochrome c can also be released from the mitochondria via Bax activation in response to various death stimuli.
Here, Bax undergoes a conformational change and consequently translocates to the mitochondria, leading to the eventual release of Cytochrome c. Cytochrome c then interacts with the pro-caspase 9 on the scaffold protein Apoptotic protease activating factor 1 (Apaf-1) and deoxyadenosine triphosphate (dATP) to form an apoptosome, leading to the activation of pro-caspase 9. This activation can initiate caspases that can then cleave and activate executioner caspases -3, -6 and -7.
Executioner caspases are capable of cleaving critical proteins and leading the cell towards apoptosis. Oligomerization and the conformational changes of Bax and/or Bak has also been shown to lead to pore formation on the outer mitochondrial membrane and thus, the subsequent release of cytochrome c and the associated downstream effects. Mitochondrial stability is therefore vital for cell viability, with disruption of mitochondria recently been associated with an increase in ROS production.
As indicated in figure 4-23, once cytochrome c has been released into the cytosol, it is capable of activating caspase 9 by interacting with the cytoplasmic protein, Apaf-1 and procaspase-9 to form the active multiprotein apoptosome. Activating caspase 9 in turn stimulates effector caspases 3, 6 and 7, resulting in degradation of cellular components and apoptosis.
Meanwhile, SMAC/DIABLO, along with HtrA2/Om, also released from the mitochondria are capable of interacting with, and impeding IAPs (inhibitors of apoptosis proteins), including XIAP (X-linked inhibitor of apoptosis protein), c-IAP1 (Apoptosis Inhibitor 1) and c-IAP2 Apoptosis Inhibitor 2)
In addition, activation of BAX/BAK also occurs at the mitochondrial outer membrane (MOM). Upon activation, BAK and BAX undergo conformational changes to form homo-oligomers and translocate to the mitochondrial membrane, disrupting mitochondrial potential and the subsequent release of important pro-apoptotic molecules such as cytochrome c and DIABLO/SMAC.
Though the exact mechanism of BAK/BAX initiation is still unknown, it has been proposed that the pro-apoptotic protein, PUMA can activate BAX and BAK-dependent permeabilization of the MOM. Although Willis et al also observed BAX/BAK activation occurring naturally once BH-3 only proteins inhibit pro-survival proteins such as Bcl-2, Bcl-xL, and Mcl-1.
The mitochondrial outer membrane (MOM) permeability is a vital catalyst to activating the intrinsic pathway, with Blc-2 members responsible for regulating its permeability. Disruption to the MOM can occur via truncated BID (tBID); the conformational change of BID to tBID is actually orchestrated by caspase-2 in response to various stimuli including an increase in ROS concentration.
The activation of tBID leads to the oligomerisation of Bak and/or Bax at the MOM, again leading to the disruption of mitochondrial outer membrane potential and the subsequent release of cytochrome c and SMAC-DIABLO into the cytosol.
The convergence between the intrinsic and extrinsic pathway is observed in this initiation as BID is also activated by caspase 8, which has been previously shown to be an active member of both the extrinsic and intrinsic pathways.
Reactive Oxygen Species
Apoptosis can also be induced by an increase in reactive oxygen species (ROS). ROS are by-products generated during oxygen metabolism within mitochondria. An overproduction of ROS can stimulate oxidative stress and damage macromolecules, proteins, lipids and DNA. Antioxidant systems help neutralise ROS generation, however a strong imbalance between antioxidants and elevated ROS concentration, can render the cell defenceless to the effects of oxidative stress.
The increased concentration of ROS in cells can cause damage to lipids, organelles, membranes and in particular, proteins and nucleic acids, further potentiating apoptosis . The intrinsic, mitochondrial, pathway is initiated by stress signals such as radiation, hypoxia, free radicals and hyperthermia.
These stimuli can directly affect the inner mitochondrial membrane, resulting in the mitochondrial permeability transition (MPT) pore, loss of the mitochondrial transmembrane potential and DNA damage. This in turn causes the release of apoptotogenic factors such as cytochrome c, apoptosis inducing factor (AIF) or Smac/DIABLO from the mitochondrial intermembrane space into the cytosol and activation of p53.
P53 is a potent tumour suppressor gene and upon activation during DNA damage or intrinsic/extrinsic signalling, acts a transcription factor to activate the transcription of target genes such as BAX, NOXA and PUMA, all key pro-apoptotic proteins involved in the disruption of the mitochondrial membrane and thus, the intrinsic pathway.
Lysosome-mediated apoptosis
For many years, caspases have been regarded as the chief instigators of apoptotic signalling however fairly recent studies have begun to show the significance of proteases from the endosomal/lysosomal systems. Lysosomes are cytoplasmic membrane-enclosed organelles containing a plethora of hydrolytic enzymes, including proteases such as cathepsin members (cathepsin -B,-D, -L) which are capable of activating pro-apoptotic effectors such as caspase 2, BID, BAX/BAK and can even directly influence the MOMP, prompting the intrinsic pathway (Boya and Kroemer 2008).
For cathepsins (and other hydrolytic enzymes) to escape the lysosome and become active within the cytosol, the lysosome membrane must first be permeabilised, this lysosomal membrane permeabilisation (LMP) can occur via various stimuli, including an increase in ROS concentration, upstream protease activation, p53 activation/DNA damage and pro-apoptotic Bcl-2 members.
LMP has also been shown to be involved in processes such as necrosis, necrotoposis, autophagy as well as apoptosis. It is thought that a quantitative relationship exists between the amounts of LMP within a cell and the pathway of cell death with more moderate stimuli triggering a modest LMP reaction leading towards more of an apoptotic pathway while more rigorous insults leading to an increased LMP response and a complete release of lysosomal contents, favouring a more necrotic pathway.
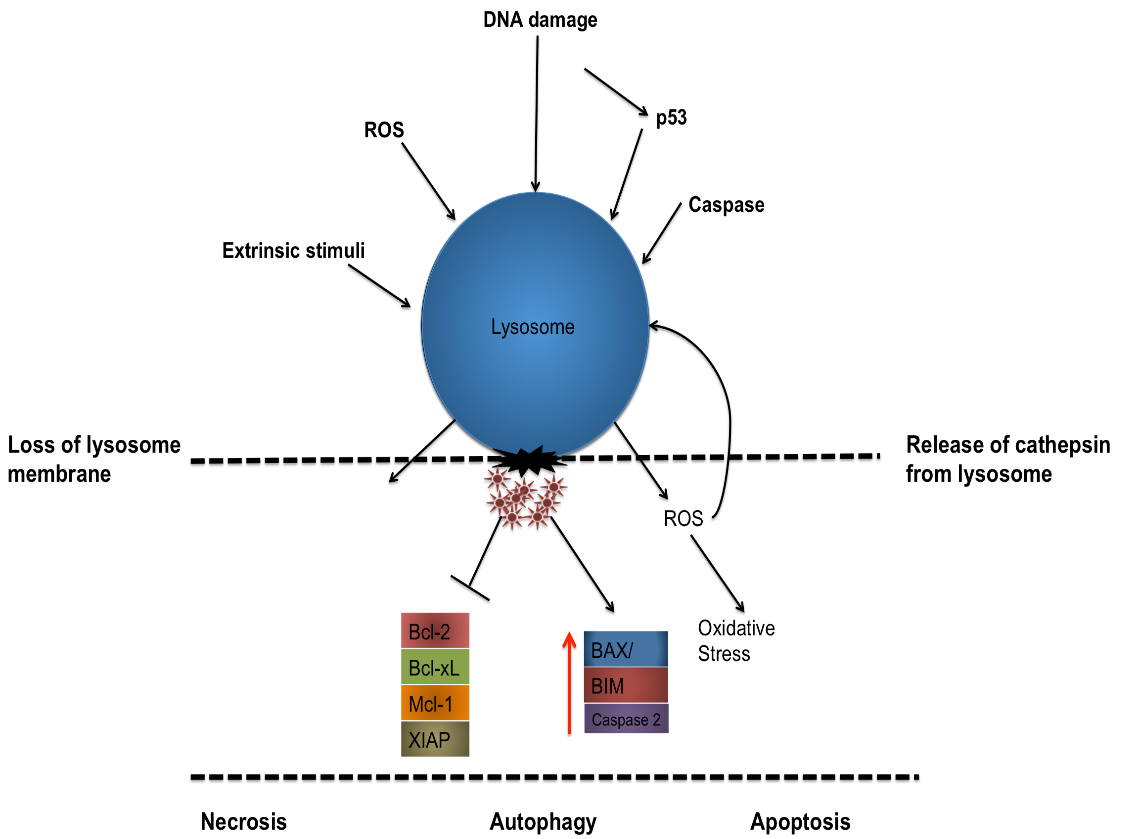
Figure 5: Schematic depicting the permeabilization of a lysosome membrane by a variety of external stimuli, DNA damage or via intracellular signalling leading to the release of cathepsin from the lysosome and the simultaneous degredation of pro-survival proteins and subsequent activation of pro-apoptotic proteins.
Summary
Many factors involved in the regulation of apoptosis.
Some factors like Fas, TNFαR, DR3, DR4, DR5 receptors, p53 proteins and caspases promote apoptosis via receptor oligomerization, while some members of the Bcl-2 family like Bcl-2, Bcl-x1, Bcl-w, Mcl-1 proteins inhibit apoptosis while the Bcl-2 family members like Bax, Bak, Bad, Bid and Bim could promote apoptosis. Bcl-2 proteins are able to inhibit or promote apoptosis by direct action on mitochondrial apoptosis-induced channel (MAC). Of all the three pathways above, caspases play an essential role in them.
They finally activate caspases which leading to apoptosis. But there also exists a caspase-independent apoptotic pathway that is mediated by apoptosis-inducing factor (AIF).
Products available
Apoptosis Assays and Kits
List of kits suitable for observing and quantifying apoptotic markers such as caspase 8,9,2. Includes MTT assay, Propidium iodide, live/dead assay, calcein A/M & ethidium homodimer TUNNEL assay and more.
Comprehensive list of Antibodies available for apoptosis
Other Apoptosis products
References:
- Adams, J. M. and Cory, S. (2007), ‘Bcl-2-regulated apoptosis: mechanism and therapeutic potential’, Curr Opin Immunol, 19 (5), 488-96.
- Aits, Sonja and Jäättelä, Marja (2013), ‘Lysosomal cell death at a glance’, Journal of Cell Science, 126 (9), 1905.
- Berghe, T. V. et al. Determination of apoptotic and necrotic cell death in vitro and in vivo. Methods 61, 117-129 (2013).
- Bonzon, C., et al. (2006), ‘Caspase-2-induced apoptosis requires bid cleavage: a physiological role for bid in heat shock-induced death’, Mol Biol Cell, 17 (5), 2150-7.
- Brenner, C. & Kroemer, G. Apoptosis. Mitochondria–the death signal integrators. Science 289, 1150-1151 (2000).
- Chun, J. N., et al. (2010), ‘Cytosolic Hsp60 is involved in the NF-kappaB-dependent survival of cancer cells via IKK regulation’, PLoS One, 5 (3), e9422.
- Du, W. and Elemento, O. (2015), ‘Cancer systems biology: embracing complexity to develop better anticancer therapeutic strategies’, Oncogene, 34 (25), 3215-25.
- Gillies, Laura A. and Kuwana, Tomomi (2014), ‘Apoptosis Regulation at the Mitochondrial Outer Membrane’, Journal of Cellular Biochemistry, 115 (4), 632-40.
- Elmore, S. (2007), ‘Apoptosis: a review of programmed cell death’, Toxicol Pathol, 35 (4), 495-516.
- Fulda, S. and Debatin, K. M. (2006), ‘Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy’, Oncogene, 25 (34), 4798-811
- Fuchs, Y. & Steller, H. Programmed cell death in animal development and disease. Cell 147, 742-758, doi:10.1016/j.cell.2011.10.033 (2011).
- Green, D. R. Means to an end : apoptosis and other cell death mechanisms. Cold Spring Harbor Laboratory (2011).
- Green, D. R. & Llambi, F. Cell Death Signaling. Cold Spring Harbor perspectives in biology 7, doi:10.1101/cshperspect.a006080 (2015).
- Kantari, Chahrazade and Walczak, Henning (2011), ‘Caspase-8 and Bid: Caught in the act between death receptors and mitochondria’, Biochimica et Biophysica Acta (BBA) – Molecular Cell Research, 1813 (4), 558-63.
- Kiraz, Y., et al. (2016a), ‘Major apoptotic mechanisms and genes involved in apoptosis’, Tumour Biol, 37 (7), 8471-86.
- 6. Krysko, D. V., Vanden Berghe, T., D’Herde, K. & Vandenabeele, P. Apoptosis and necrosis: detection, discrimination and phagocytosis. Methods 44, 205-221, doi:10.1016/j.ymeth.2007.12.001 (2008).
- McIlwain, David R., Berger, Thorsten, and Mak, Tak W. (2013b), ‘Caspase Functions in Cell Death and Disease’, Cold Spring Harbor Perspectives in Biology, 5 (4), a008656.
- Ilmarinen, Pinja, Moilanen, Eeva, and Kankaanranta, Hannu (2014), ‘Mitochondria in the Center of Human Eosinophil Apoptosis and Survival’, International Journal of Molecular Sciences, 15 (3), 3952-69.
- Ivanova, Saka, et al. (2008), ‘Chapter Nine Lysosomes in Apoptosis’, Methods in Enzymology (Volume 442: Academic Press), 183-99.
- Jolesch, A., et al. (2012), ‘Hsp70, a messenger from hyperthermia for the immune system’, Eur J Cell Biol, 91 (1), 48-52.
- Jolly, C. and Morimoto, R. I. (2000), ‘Role of the heat shock response and molecular chaperones in oncogenesis and cell death’, J Natl Cancer Inst, 92 (19), 1564-72.
- Martinez-Ruiz, G., et al. (2008a), ‘Role of Smac/DIABLO in cancer progression’, J Exp Clin Cancer Res, 27, 48.
- Martinez-Ruiz, Gustavo, et al. (2008b), ‘Role of Smac/DIABLO in cancer progression’, Journal of Experimental & Clinical Cancer Research : CR, 27 (1), 48-48.
- Nakano, Katsunori and Vousden, Karen H. (2001), ‘PUMA, a Novel Proapoptotic Gene, Is Induced by p53’, Molecular Cell, 7 (3), 683-94.
- Pagliari, L. J., et al. (2005), ‘The multidomain proapoptotic molecules Bax and Bak are directly activated by heat’, Proc Natl Acad Sci U S A, 102 (50), 17975-80.
- Ott, M., et al. (2007), ‘Mitochondria, oxidative stress and cell death’, Apoptosis, 12 (5), 913-22.
- Ou, J. R., et al. (2014), ‘Heat shock protein 90 in Alzheimer’s disease’, Biomed Res Int, 2014, 796869.
- Repnik, Urška, Česen, Maruša Hafner, and Turk, Boris (2013), ‘The Endolysosomal System in Cell Death and Survival’, Cold Spring Harbor Perspectives in Biology, 5 (1).
- Ricci, Jean-Ehrland, et al. (2004), ‘Disruption of Mitochondrial Function during Apoptosis Is Mediated by Caspase Cleavage of the p75 Subunit of Complex I of the Electron Transport Chain’, Cell, 117 (6), 773-86.
- Saelens, X., et al. (2004), ‘Toxic proteins released from mitochondria in cell death’, Oncogene, 23 (16), 2861-74.
- Susin, S. A. et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441-446, doi:10.1038/17135 (1999).
- Slimen, I. B., et al. (2014), ‘Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review’, Int J Hyperthermia, 30 (7), 513-23.
- Wang, Chunxin and Youle, Richard J. (2009), ‘The Role of Mitochondria in Apoptosis()’, Annual review of genetics, 43, 95-118
- Wang, Kewei and Lin, Bingliang (2013), ‘Inhibitor of apoptosis proteins (IAPs) as regulatory factors of hepatic apoptosis’, Cellular Signalling, 25 (10), 1970-80
- Wang, Z., et al. (2013c), ‘The role of mitochondria-derived reactive oxygen species in hyperthermia-induced platelet apoptosis’, PLoS One, 8 (9), e75044.
- Willis, Simon N., et al. (2007), ‘Apoptosis Initiated When BH3 Ligands Engage Multiple Bcl-2 Homologs, Not Bax or Bak’, Science, 315 (5813), 856.
- Yan, Biao (2011), ‘Research progress on Livin protein: an inhibitor of apoptosis’, Molecular and Cellular Biochemistry, 357 (1), 39-45.
- Yang, Qi-Heng and Du, Chunying (2004), ‘Smac/DIABLO Selectively Reduces the Levels of c-IAP1 and c-IAP2 but Not That of XIAP and Livin in HeLa Cells’, Journal of Biological Chemistry, 279 (17), 16963-70.
- Yang, X., et al. (2012a), ‘Hsp70 promotes chemoresistance by blocking Bax mitochondrial translocation in ovarian cancer cells’, Cancer Lett, 321 (2), 137-43.
- Yang, Xiaokui, et al. (2012b), ‘Hsp70 promotes chemoresistance by blocking Bax mitochondrial translocation in ovarian cancer cells’, Cancer Letters, 321 (2), 137-43.
- Zheng, J. H., et al. (2015), ‘Discoveries and controversies in BCL-2 protein-mediated apoptosis’, FEBS J.
- Zheng, Janet H., et al. (2016), ‘Discoveries and controversies in BCL-2 protein-mediated apoptosis’, The FEBS Journal, 283 (14), 2690-700.