What is Autophagy?
Autophagy (namely self-eating) is a highly conserved process for disassembling intracellular excess and dysfunctional components such as misfolded/aggregated proteins, damaged organelles and invading pathogens. Autophagy therefore plays an integral role in the maintenance of cellular homeostasis, differentiation and development, immunity and in eliminating diseases.
Autophagy involves a catabolic process in the degradation of cells; helping remove dysfunctional cellular components via lysosome-mediated degradation. By preferentially targeting substrates that are damaged or dysfunctional, autophagy can contribute to the quality control and replacement of cellular component during damage/cellular stress. Organelles to be degraded are surrounded by a double membrane, which then forms a structure known as an autophagosome. The autophagosome combines with lysosomes and forms what is referred to as the autolysosome.
Currently, the major characterised types of autophagy include macro-autophagy, micro-autophagy, and chaperone-mediated autophagy. Autophagy is strictly regulated allowing cells recover and remain viable in response to multiple toxic stimuli. Three commonly known variants of autophagy exists including macroautophagy, microautophagy, and chaperone-mediated autophagy, but the term “autophagy” usually indicates macroautophagy unless otherwise specified. Below, we have summarised these types.
Forms of Cell Autophagy
Macro-autophagy is the major type of autophagy. It involves the sequestering of cellular constituents in double-membrane vesicles (autophagosomes) and subsequent delivery to lysosomes for degradation.
It can be nonselective or selective. Nonselective autophagy is used for the turnover of bulk cytoplasm under starvation conditions. Selective autophagy targets damaged or superfluous mitochondria (mitophagy), peroxisomes (pexophagy), lipid droplets (lipophagy) and microbes (xenophagy).
Micro-autophagy is the second type of autophagy. The lysosome itself engulfs small components of the cytoplasm by inward invagination of the lysosomal membrane.
Chaperone-mediated autophagy does not involve membrane reorganization, instead, substrate proteins directly translocate across the lysosomal membrane during chaperone-mediated autophagy.
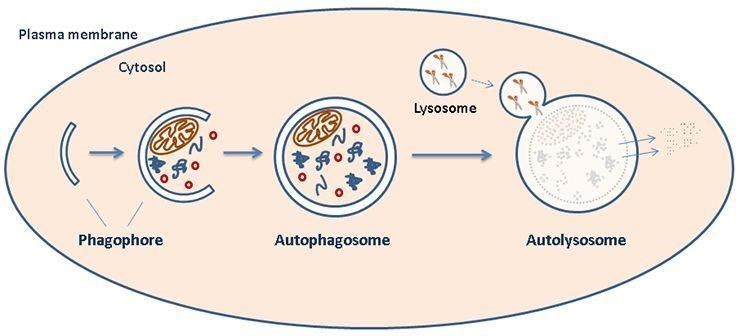
Figure 1: schematic representation of organelle degradation.
Process and regulation of autophagy
Autophagy is mainly regulated by nutrients, growth factors and stress. It begins with the formation of the ‘‘isolation membrane’’ also known as phagophore which then extends and closes to become autophagosomes. Subsequently, autophagosomes mature by fusing with late endosomes and lysosomes, thereby forming autolysosomes.
In mammalian cells, Protein kinase B (PKB, also known as Akt), mitogen-activated protein kinase (MAKP)-Erk, p53, and adenosine 5’ adenosine monophosphate-activated protein kinase (AMPK) pathways participate in the expression of autophagy-related genes (Atgs).
Autophagy is controlled by the orchestration of more than 30 Atgs whose expression is induced by FoxO. FoxO1 tends to induce Atg12 expression and FoxO3 priors to activating Lc3 genes. It has been reported that JNK is an effective negatively regulator of FoxO-dependent autophagy in neurons. The anti-apoptotic protein Bcl2 and other Bcl family members can inhibit autophagy.
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase that belongs to the phosphatidylinositol kinase-related kinase (PIKK) family. It was first described as the physiological target of the immunosuppressant drug rapamycin.
Subsequent studies established its role in protein translation and cell growth. Owing to its energy sensing functions, mTOR is considered as the master regulator of autophagy. The mTOR kinase is activated downstream of Akt kinase, MAKP-Erk and growth factor receptor, signaling when nutrients are available and acting to promote growth through induction of ribosomal protein expression and increased protein translation.
While mTOR kinase is negatively regulated by p53 and AMPK pathway. The UNC-51-like kinase (ULK) acts downstream of the mTOR complex. ULK1 and ULK2 form a large complex, which is also the formation of phagophore, with the mammalian homolog of Vac8, Atg13, Atg11, and the scaffold protein FIP200.
Class III PI3K complex, containing Atg13, Atg9, hVps34, and ultraviolet irradiation resistance-associated gene (UVRAG), is required for the induction of autophagy. The Atgs control autophagosome formation through a large complex of Atg12, Atg5 and Atg16L.
Atg12 is conjugated to Atg7, Atg10, Atg5, and Atg16L. The LC3 is cleaved at its C-terminus by Atg4 protease to generate the cytosolic LC3-I which is conjugated to phosphatidylethanolamine (PE) also in a ubiquitin-like reaction that requires Atg7 and Atg3. The lipidated form of LC3, known as LC3-II, is attached to the autophagosome membrane.
Autophagy and apoptosis are connected both positively and negatively, and extensive crosstalk exists between the two processes. LC3-II interact with Rab which could be the candidate for lysosome biogenesis, and then induced by vesicle-associated membrane protein (VAMP8), synaptosomal-associated protein 29 (SNAP29), and syntaxin-17 (SNX17) to participate in autophagolysosome.
The MAPK signal transduction pathway is one of the most important regulatory mechanisms in eukaryotic cells, and MAPK sub-families JNK participates in multiple stimulation-induced autophagic events, including endoplasmic reticulum stress, caspase (cysteine aspartate-special proteases) inhibition, insulin-like growth factor-1 treatment and exposure to tumor necrosis factor- α (TNF-α).
Moreover, JNK has been related to autophagicinduced cell death. The concept of autophagic cell death is redefined as a modality of non-apoptotic or necrotic programmed cell death (PCD) in which autophagy serves as a cell death mechanism. When cells act to down-regulate oncogenic gene expression, oncogenic stress can trigger autophagic cell death as a defense mechanism. Activated JNK induces FoxOs nuclear localization and increases its activity to regulate transcription of other ‘core’ ATG genes.
The pathways of Autophagy
Autophagy consists of several sequential steps including sequestration, transport to lysosomes, degradation, and utilization of degradation products. Autophagy is initiated with the production of the autophagosome, a double-membrane intracellular structure of reticular origin that engulfs targeted cytoplasmic contents and ultimately fuses with lysosomes for eventual degradation.
Macroautophagy (referred to throughout this article as autophagy), is the main pathway which is primarily used to eradicate damaged organelles or unused proteins and can be further divided into either canonical or non-canonical autophagy. In this article, we focus on the research progress of macroautophagy.
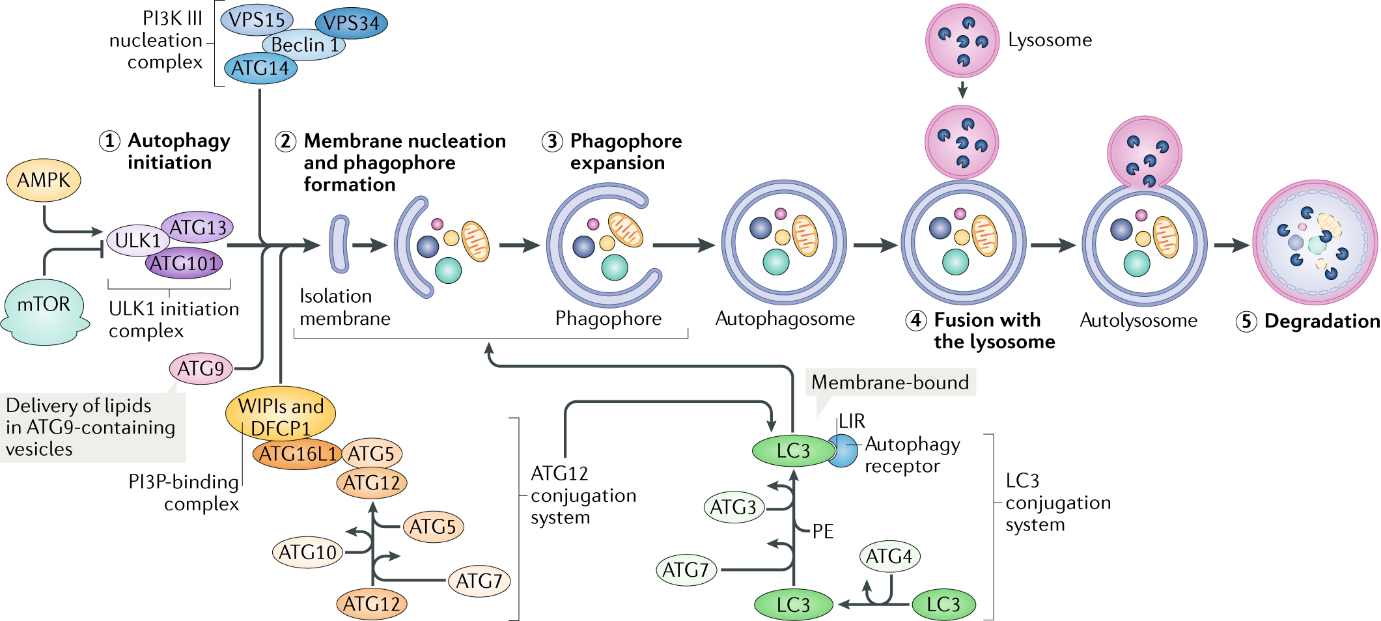
Figure 2: overview of autophagy( https://www.nature.com/articles/s41580-018-0033-y)
- Canonical Autophagy
The canonical autophagy pathway is largely considered a non-specific process, and is a highly conserved process by which eukaryotic cells scavenge their own cytoplasmic contents via a double membrane that encloses and isolates the cytoplasmic components into a phagophore. Phagophores then fuses with lysosomes for degradation.
MTOR is the key regulator of the molecular mechanisms of canonical autophagy. Autophagy-inducing stimulus (such as nutrient deprivation, stress, hypoxia) triggers the activation of AMPK, whose kinase activity inhibits mTOR, and then activates the pre-initiation complex, composed of ULK1/2, ATG13, ATG101 and FIP200. This complex then activates the PI3K complex(VPS34, Beclin 1 and ATG14). The PI3K complex produces phosphatidylinositol 3-phosphate (PI3P), which acts as recruitment messenger for the downstream two ubiquitin-like conjugation systems.
One of ubiquitin-like conjugation systems is responsible for the formation of a supramolecular protein complex containing ATG5, ATG12 and autophagy-related 16-like 1 (ATG16L1), including autophagy-related 7 (ATG7) and ATG10, another composes of ATG3, ATG4 and ATG7, promotes the cleavage of members of the Atg8 protein family, including human LC3, and their conjugation to phosphatidylethanolamine (PE).
The activity and coordination of these two systems facilitate the expansion and sealing of the autophagosome, as well as the lipidation and embedding of LC3-PE into the autophagosomal membrane/
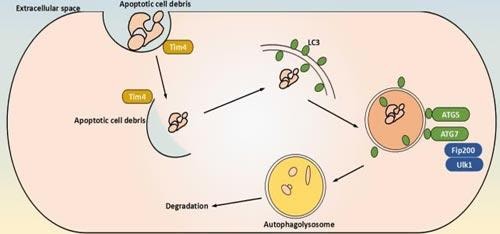
Figure 1: schematical representation of canonical autophagy
- Non-canonical Autophagy
The non-canonical autophagy is considered a specific process that selectively targets internal cellular substrates such as organelles for autophagy. Some of these processes include ribophagy , pexophagy, lipophagy, chlorophagy, and mitophagy.
Currently, mitophagy is a representative of selective autophagy, which can be regulated by several different mechanisms depending on the physiological context. Here we present a short overview of two different mechanisms that regulate mitophagy.
One key regulator is Parkin. Upon damage or depolarization, the mitochondrial kinase PteN-induced kinase 1 (PiNK1) becomes stabilized and recruits the Ub E3 protein ligase Parkin. PiNK1 and Parkin assemble phosphorylated Ub (pUb) chains on several proteins of the outer mitochondrial membrane via a feedforward mechanism.
These proteins of the outer mitochondrial membrane in turn recruit cargo receptors such as calcium-binding and coiled-coil domaincontaining protein 2 (NDP52) and optineurin (OPtN). In this process, free Ub is phosphorylated by PiNK1, and Parkin attaches polyUb to the mitochondrial surface and the ubiquitin-like (uBl) domain of Parkin. these phosphorylation events enhance both the ubiquitin ligase activity of Parkin and its retention time on damaged mitochondria.
Another player in mitophagy is taNKbinding kinase 1 (tBK1), which promotes coupling of the cargo to the phagophore via phosphorylating ub-binding domains and LIRs of several cargo receptors, thereby increasing their affinity for pub and LC3, respectively.
Notably, mitophagy can also occur in a ub-independent manner via mitochondrial proteins such as BCL2/adenovirus E1B protein-interacting protein 3-like (NiX), FuN14 domain-containing protein 1 (FuNDC1) and BCL2/adenovirus E1B protein-interacting protein 3 (BNiP3), which possess an LC3-interacting region (LIR) and therefore function as direct cargo receptors; they are typically regulated by stress-dependent phosphorylation. Finally, lipids, including phospholipids, such as cardiolipin and ceramide, have been shown to mediate mitophagy in neuronal cells.
Cardiolipin is located at the inner membrane of healthy mitochondria, but upon mitochondrial damage, it is externalised and presented on the mitochondrial surface, where it is recognised by LC3.
Core autophagy proteins of the autophagic pathway
As previously mentioned, autophagy is a cellular catabolic pathway involving in protein degradation, organelle turnover, and non-selective breakdown of cytoplasmic components. This progress consists of six stages, including induction, phagophore nucleation, phagophore expansion, autophagosome formation, lysosome fusion, and component degradation. The core proteins of these stages are shown in table 1.
Table 1. Key autophagic factors and their regulation (adapted from Ivan Dikic, 2018)
| Protein | Function | Mechanisms of regulation |
| Initiation and phagophore nucleation | ||
| ULK1 and ATG1 | Serine/threonine kinase; initiates autophagy by phosphorylating components of the autophagy machinery | Stress and nutrients (via mTORC1, AMPK and LKB1); TFEB and several miRNAs |
| FIP200 | Component of ULK complex (possibly scaffolding function) | ULK1 and miRNAs |
| ATG13 | Adaptor mediating the interaction between ULK1 and FIP200; enhances ULK1 kinase activity | ULK1, mTORC1 and AMPK |
| ATG101 | Component of ULK complex; recruitment of downstream ATG proteins | ULK1 |
| VPS34 | Catalytic component of PI3KC3–C1; generates PI3P in the phagophore and stabilizes the ULK complex | AMPK, ULK1 and p300 (acetylation) |
| Beclin 1 | Promotes formation of PI3KC3–C1 and regulates the lipid kinase VPS34 | Activation: AMPK, ULK1, MAPKAPK2, MAPKAPK3, DAPK and UVRAG; inhibition: BCL-2, AKT and EGFR |
| ATG14 | PI3KC3–C1 targeting to the PAS and expanding phagophore | PIPKIγI5 and mTORC1 |
| ATG9 | Delivery of membrane material to the phagophore | ULK1 complex |
| WIPI2 | PI3P-binding protein that recruits ATG12-ATG5-ATG16L to the phagophore; retrieval of ATG9 from early autophagosomal membranes | TFEB (positive transcription regulator) and ZKSCAN3 (negative transcription regulator) |
| Phagophore expansion | ||
| ATG4 | Cysteine protease that processes pro-ATG8s; also, deconjugation of lipidated LC3 and ATG8s | ULK1 and ROS |
| ATG7 | E1-like enzyme; activation of ATG8; conjugation of ATG12 to ATG5 | miRNAs |
| ATG3 | E2-like enzyme; conjugation of activated ATG8s to membranal PE | miRNAs |
| ATG10 | E2-like enzyme that conjugates ATG12 to ATG5 | miRNAs |
| ATG12~ATG5–ATG16L | E3-like complex that couples ATG8s to PE | CSNK2 |
| PE-conjugated ATG8s | Scaffold for assembly of the ULK1 complex; supports membrane tethering and hemifusion events for phagophore expansion | ULK1, PKA, ATG4 and mTOR |
| ATG9 | Delivery of membrane material to the phagophore | ULK1 |
| Autophagosome formation | ||
| Ubiquitin | Cargo labelling | PINK (phosphorylation) |
| Cardiolipin and ceramide | Cargo labelling | Phosphorylation |
| p62 | Autophagy receptor | ULK1 and TBK1 |
| OPTN | Autophagy receptor | TBK1 |
| NBR1 | Autophagy receptor | TBK1 |
| NDP52 | Autophagy receptor | TBK1 |
| PE-conjugated LC3 | Interaction with autophagy receptors; also phagophore expansion and sealing | ULK1, PKA, ATG4 and mTOR |
| LC3s and GABARAPs | Unclear | Unclear; might involve phosphorylation and acetylation events |
| ATG4 | Removal of ATG8s from the surface of the autophagosome | Unknown |
| PE-conjugated LC3s and GABARAPs | Linking the autophagosome to microtubule-based kinesin motor | Unclear; might involve phosphorylation and acetylation events |
| Fusion with lysosome | ||
| PE-conjugated LC3s and GABARAPs | Mediates autophagosome–lysosome fusion upon phosphorylation through PLEKHM1 and HOPS | STK3 and STK4 |
| ATG14 | Promotes SNARE-driven membrane fusion | Unknown |
| Rab GTPase RAB7 | Unclear | Unknown |
Summary
Autophagy is a fundamental biological process by removing damaged organelles, but disordered autophagy is involved in a variety of diseases including neurodegeneration and microbial infection. Autophagy is activated in response to adverse environmental conditions such as the deprivation of nutrients, hypoxia, pathogen infection, radiation and oxidative stress as a survival mechanism.
This process plays a role in cellular homeostasis, development, and longevity and has many effects on the cellular renovation. It would be reasonable to assume that autophagy can contribute to whole-body rejuvenation. Under many conditions, autophagy is considered as a physiologic cytoprotective or pro-survival mechanism, however, completely uncontrolled or excessive autophagy has been associated with cell death.
The characterization of the regulation of autophagy has become relevant because defective autophagy has been linked to aging, neurodegenerative disorders.
Products available
Antibodies for prominent autophagy markers
Bioss Autophagy Markers Antibodies
Autophagy Antibodies from Stratech
Autophagy Assay & kits
Inhibitors
ELISA kits
References:
- Amaravadi, R., Kimmelman, et al. Recent insights into the function of autophagy in cancer[J]. Genes Dev. 2016, 30, 1913–1930.
- An, Heeseon, Harper, J. Wade. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy[J]. Nature Cell Biology. 2018, 20 (2): 135–143.
- Budina-Kolomets, A., et al. (2014), ‘Comparison of the activity of three different HSP70 inhibitors on apoptosis, cell cycle arrest, autophagy inhibition, and HSP90 inhibition’, Cancer Biol Ther, 15 (2), 194-9.
- Chu, C. T. et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells[J]. Nat. Cell Biol. 2013,15, 1197–1205.
- Chen, Kuo-Li, et al. (2012), ‘Targeting cathepsin S induces tumor cell autophagy via the EGFR–ERK signaling pathway’, Cancer Letters, 317 (1), 89-98.
- Dokladny, Karol, Myers, Orrin B., and Moseley, Pope L. (2015), ‘Heat shock response and autophagy—cooperation and control’, Autophagy, 11 (2), 200-13.
- Eskelinen, Eeva-Liisa and Saftig, Paul (2009), ‘Autophagy: A lysosomal degradation pathway with a central role in health and disease’, Biochimica et Biophysica Acta (BBA) – Molecular Cell Research, 1793 (4), 664-73.
- Galluzzi, L. Autophagy in malignant transformation and cancer progression[J]. EMBO J. 2015.
- Ding, Wen-Xing; Yin, et al. Mitophagy: mechanisms, pathophysiological roles, and analysis[J]. Biological Chemistry. 2012, 393 (7).
- Ivan Dikic, and Zvulun Elazar. Mechanism and medical implications of mammalian autophagy[J]. Nat. Rev. Mol. Cell Biol. 2018.
- Jean M. Mulcahy Levy, Christina G. Towers, et al. Targeting autophagy in cancer[J]. Nat Rev Cancer. 2017, 17(9):528-542.
- Kroemer, Guido and Jaattela, Marja (2005), ‘Lysosomes and autophagy in cell death control’, Nat Rev Cancer, 5 (11), 886-97.
- Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy[J]. Cell Death and Differentiation. 2013, 20 (1): 3–11.
- Mariño, Guillermo, et al. (2014), ‘Self-consumption: the interplay of autophagy and apoptosis’, Nature reviews. Molecular cell biology, 15 (2), 81-94.
- Nikoletopoulou, Vassiliki, et al. (2013), ‘Crosstalk between apoptosis, necrosis and autophagy’, Biochimica et Biophysica Acta (BBA) – Molecular Cell Research, 1833 (12), 3448-59.
- [1] Noda, N. N. & Inagaki, F. Mechanisms of autophagy. Annu[J]. Rev. Biophys. 2015, 44, 101-122.
- Lorenzo Galluzzi1, José Manuel Bravo-San Pedro, et al. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles[J]. Nat Rev Drug Discov. 2017 16(7):487-511.
- Mizushima N, Yoshimori T, et al. The role of Atg proteins in autophagosome formation[J]. Annual Review of Cell and Developmental Biology. 2011, 27 (1): 107-32.
- Mizushima N, Ohsumi Y, et al. Autophagosome formation in mammalian cells[J]. Cell Structure and Function. 2002, 27 (6): 421-9.
- Petrache Voicu, Sorina Nicoleta, et al. (2015), ‘Silica Nanoparticles Induce Oxidative Stress and Autophagy but Not Apoptosis in the MRC-5 Cell Line’, International Journal of Molecular Sciences, 16 (12), 29398-416.
- Rodríguez-Vargas, José Manuel, et al. (2012), ‘ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy’, Cell Research, 22 (7), 1181-98.
- Safa, Ahmad R. (2013), ‘Roles of c-FLIP in Apoptosis, Necroptosis, and Autophagy’, Journal of carcinogenesis & mutagenesis, Suppl 6, 003.
- Sentelle, R. D. et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy[J]. Nat. Chem. Biol. 2012, 8, 831–838
- Till, Andreas, Lakhani, Ronak, et al. Pexophagy: The Selective Degradation of Peroxisomes[J]. International Journal of Cell Biology. 2012: 1–18.
- White, E. Deconvoluting the context-dependent role for autophagy in cancer[J]. Nat. Rev. Cancer. 2012, 12, 401–410 .
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations[J]. Nature Cell Biology. 2007, 9 (10): 1102-9.
- Yang, Qi-Heng and Du, Chunying (2004), ‘Smac/DIABLO Selectively Reduces the Levels of c-IAP1 and c-IAP2 but Not That of XIAP and Livin in HeLa Cells’, Journal of Biological Chemistry, 279 (17), 16963-70.
- Yang, X., et al. (2012a), ‘Hsp70 promotes chemoresistance by blocking Bax mitochondrial translocation in ovarian cancer cells’, Cancer Lett, 321 (2), 137-43.
- Yang, Xiaokui, et al. (2012b), ‘Hsp70 promotes chemoresistance by blocking Bax mitochondrial translocation in ovarian cancer cells’, Cancer Letters, 321 (2), 137-43.
- Zhang, Yue and Calderwood, Stuart K. (2011), ‘Autophagy, Protein Aggregation and Hyperthermia: A Minireview’, International journal of hyperthermia : the official journal of European Society for Hyperthermic Oncology, North American Hyperthermia Group, 27 (5), 409-14.
- Zhou, Feifan, Yang, Ying, and Xing, Da (2011), ‘Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis’, FEBS Journal, 278 (3), 403.