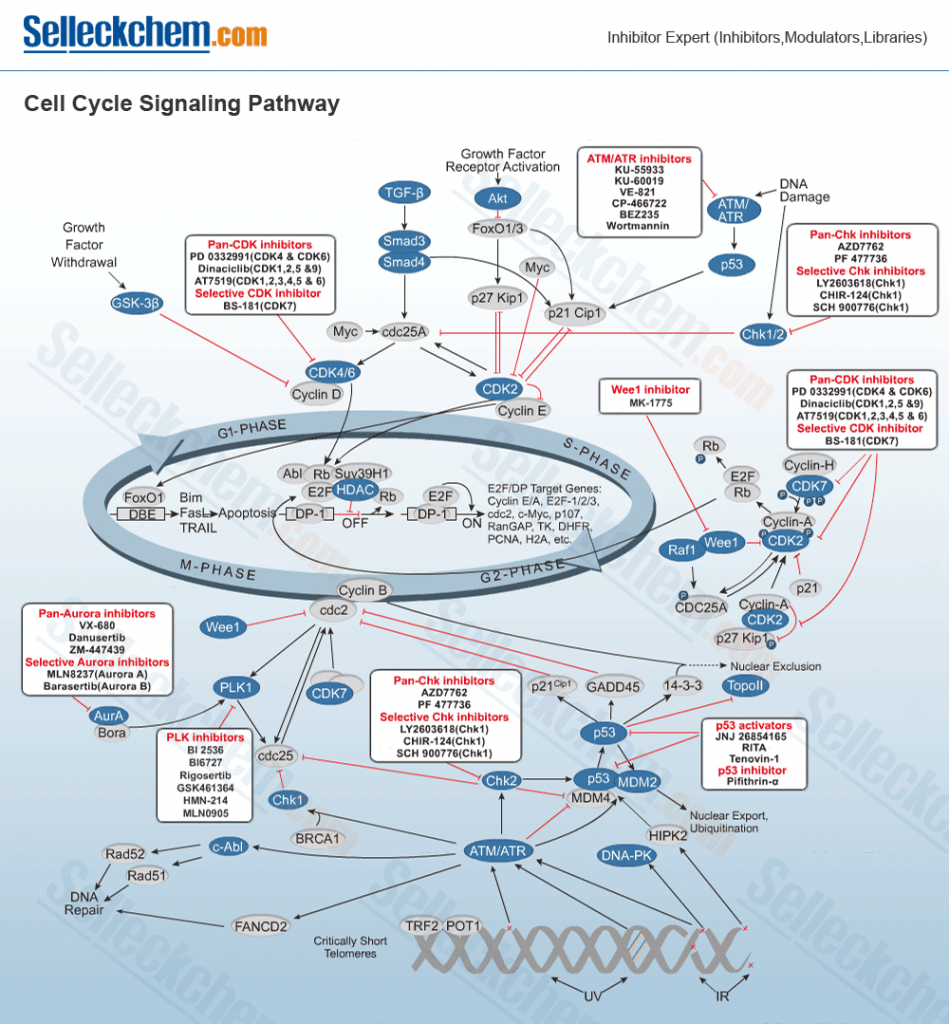
The cell cycle and cell cycle-associated proteins play essential roles in cell fate, including cell replication, cell death, and cell function. Cell cycle-check points include many targets such as CDK, ROCK, Topoisomerase, Antimetabolites and Microtubule.
Selleck Chemicals supply products to the following targets:
- Aurora Kinase
- CDK
- PLK
- Chk
- ROCK
- APC
- C-Myc
- Rac
- Wee1
There are five stages of the cell cycle:
- S-phase, when DNA replication occurs.
- G1 phase follows from mitosis and is a period when the cell is sensitive to positive and negative cues from growth signalling networks.
- G2 phase, when the cell prepares for entry into mitosis.
- M phase, when the cell divides into two daughter cells
- G0, this is a time when cells have reversibly withdrawn from the cell division cycle in response to high cell density or mitogen deprivation. Terminal differentiation or senescent out-of-cycle states are possible as well as leading to cells that are irreversibly withdrawn from the cell cycle.
Maintaining a complete and undamaged genome in an environment characterised by various potential sources of DNA integrity is essential to cell survival and reproduction. With the exception of embryonic cells, the cell cycle in eukaryotes contains checkpoints to monitor all cell cycle events involving DNA metabolism. The supervision of DNA replication not only occurs in the S-phase of cell division, but throughout the entire cell cycle process to minimise damage accruement by halting cells when the DNA is compromised and to mediate its repair. The three major checkpoint pathways are defined by a grouping of proteins that act in concert to translate the signal of damaged DNA into the response of cell cycle arrest and repair. These groups include; 1) sensor proteins, that recognise damaged DNA directly or indirectly and function to signal the presence of abnormalities, initiating a biochemical cascade of activity, 2) transducer proteins, typically protein kinases, that relay and amplify the damage signal from the sensors by phosphorylating other kinases or downstream target proteins and 3) effector proteins, which include the most downstream targets of the transducer protein kinases, and are thus regulated, usually by phosphorylation, to prevent cell cycle progression. [1]
Sensor proteins have been grouped together based on genetic and/or biochemical findings or inference that has illustrated their ability to associate with damaged DNA, either directly or indirectly. The sensor proteins that associate with a particular DNA lesion may serve as recognition complexes to modulate, recruit, and localise specific target proteins for a particular lesion. In mammals, there are several proteins that have been identified to have a sensory role, including: Rad17, Rad9, Rad1, Hus1, BRCA1, TopBP1, Mre11, Rad50, Nbs1, TopBP1, Pol2, BLM, WRN, Top3, Rfc2-5, Rpa2, PCNA, and 9-1-1. [1][2]
Transducers are protein kinases that are activated by DNA damage to initiate a signal transduction cascade to propagate and amplify communication signals that indicate damage. The result of transducer activity is to cause cell cycle arrest. In mammals, transducer proteins include Mec1, Tel1, ATR, ATM, ATRIP, Chk1, and Chk2/Rad53.
Effector target proteins include Dun1 and Cdk1 protein kinases, Cdc45, p53 transcription factor, MCM helicase, and Pol? that result in cell cycle arrest, transcription, DNA repair, and trans-lesion synthesis. [2]
The importance of DNA checkpoints has long been recognised in the field of oncology. For instance, the p53 transcription factor plays a significant role in arresting the G1 cell cycle and inducing apoptosis following DNA damage. It is known that abnormal p53 functioning contributes to tumorigenesis. However, the understanding of how p53 functions to arrest G1 and G2 cell cycles and cause apoptotic events remains unclear at the molecular level.
Another key checkpoint agent is the ataxia telangiectasia mutated (ATM) kinase that plays a pivotal role in all DNA maintenance responses, especially when double-stranded breaks have occurred.[1] ATM activation is necessary for checkpoint activation, which is dependent on the autophosphorylation of Ser1981 and the interaction with the Mre11-Rad50-Nbs1 (MRN) complex at the double strand break (DSB) sites. However, it remains unclear which DNA structures stimulate ATM activation. In unperturbed cells, ATM is an inactive dimer. In response to DSB’s, ATM is quickly autophosphorylated at Ser1981, which coincides with ATM forming active monomers. Aside from Ser1981, ATM is autophosphorylated at Ser367 and Ser1893. Cells harboring mutations in these sites are more sensitive to DSB’s and show defective checkpoint signalling. As a transducer, ATM activates multiple proteins in a special protein complex to achieve its objectives, including the activation of p53 protein and Chk2, with some crosstalk with Chk1. Activated Chk2 phosphorylates p53 and prevents Mdm2 from binding with p53, leading to its stabilisation. Cell cycle arrest or apoptosis in the G1 phase can then be directed by activated p53. ATM can also phosphorylate BRCA1, and its activation can have important functions related to managing DNA damage response. [3]
The ataxia telangiectasia and Rad3-related (ATR) kinase is another important transducer in cell cycle checkpoints. ATR signalling is initiated after DNA damage in response to RPA that is loaded onto single-stranded DNA (ssDNA) gaps or resected DSB’s. The molecule shares some sequence homology with ATM, but the molecule shows preference towards Chk1 as a substrate. In contrast, ATM is primarily targeted by Chk2. In addition, there is a high degree of redundancy in DNA damage cell cycle pathways initiated by either ATM or ATR that leads to the activation of Cdc25 phosphatases (Cdc25A, Cdc25B, and Cdc25C). Cdc25A controls progression through S-phase and mitosis, while Cdc25B and Cdc25C control mitotic entry. Overexpression of Cdc25 phosphatases is observed in a number of different human cancers. [4]
In addition to ATM and ATR, the mammalian cell cycle is regulated by numerous Cdk (cyclin-dependent kinase)-cyclin complexes at the cell cycle checkpoints. In the early G1 phase of the cell cycle, Cdk4/6-Cyclin D complexes are active. Subsequently, entry into and progression through S-phase are regulated by Cdk2-Cycline E and Cdk2-Cyclin A, respectively, while the onset of mitosis is managed by Cdk1 (Cdc2)-Cyclin B. In addition, the activities of Cdk’s are inhibited by numerous cyclin-dependent kinase inhibitors, such as p21. [3]
Currently, there are a number of cell cycle inhibitors that are undergoing human clinical evaluation. These inhibitors can be classified into the following molecular classes: Pyrazines, Pyrrolo/pyrazolopyridines, Tricyclics, Ortho-Diaminoacryls, Five-membered heterocyclic amides/amidines, and Imidazo[1,2-a]pyrazines and pyrazolo [1,5-a]pyrimidines. [4]
References
- Nyberg, KA, Michelson, RJ, Putnam, CW and Weinert, TA. Toward Maintaining the Genome: DNA Damage and Replication Checkpoints. Annu. Rev. Genet. 2002: 36; 617–56.?
- [2] Sclafani, RA and Holzen, TM. Cell Cycle Regulation of DNA Replication. Annu. Rev. Genet. 2007: 41; 237–80.
- Poehlmann, A and Roessner, A. Review. Importance of DNA Damage Checkpoints in the Pathogenesis of Human Cancers. Pathology – Research and Practice. 2010: 206; 591-601.?
- Lainchbury, M. and Collins, I. Checkpoint kinase inhibitors: a patent review (2009-2010). Expert Opin. Ther. Patents. 2011: 21(8); 1191-1210.