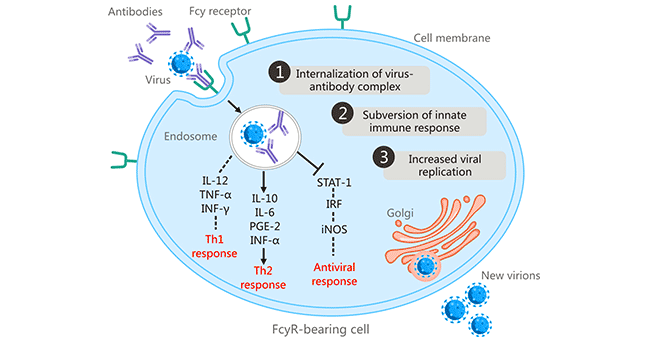
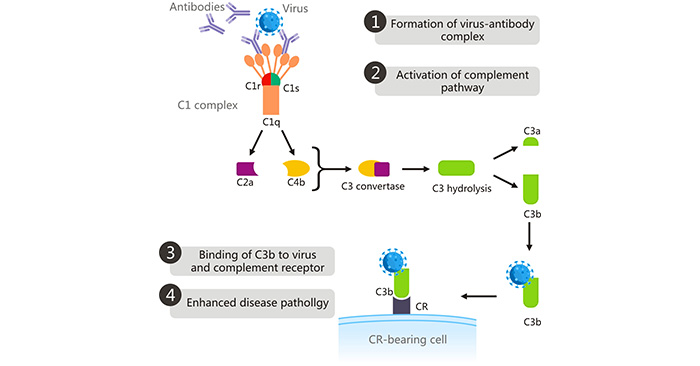
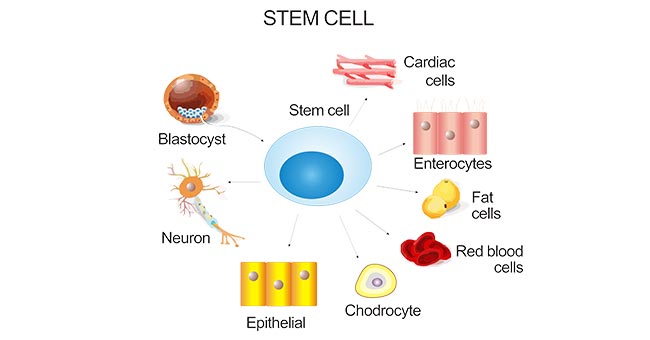
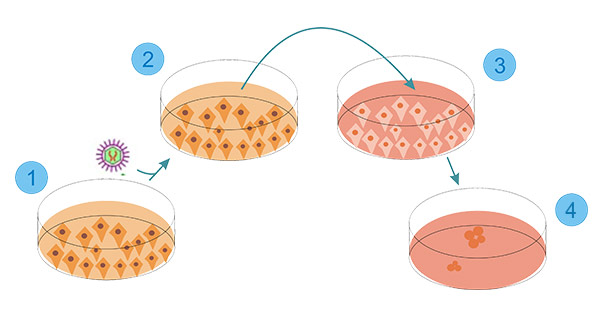
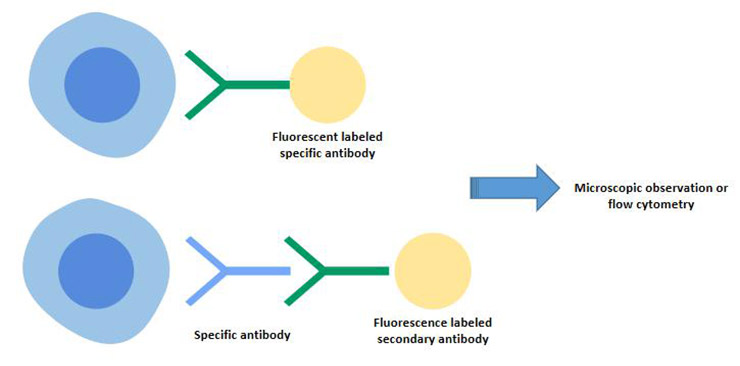
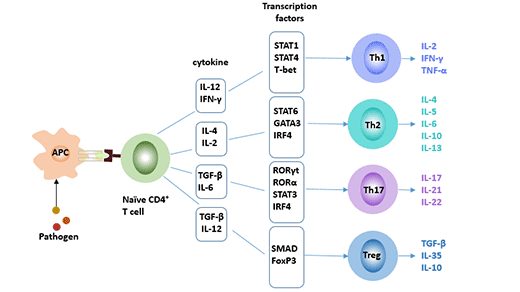
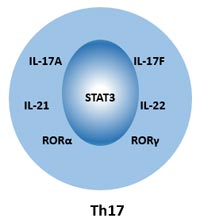
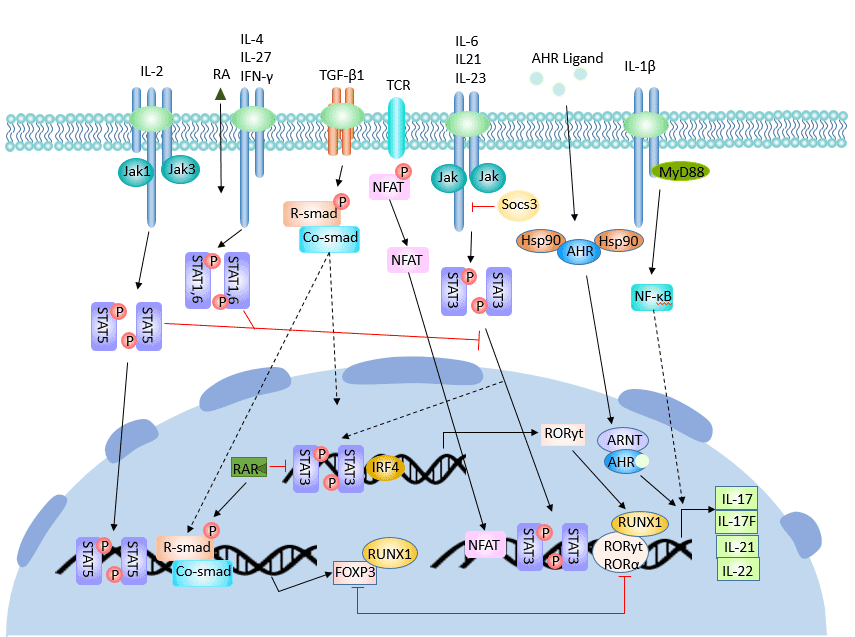
[1] Stauber DJ, DiGabriele AD, et al. Structural interactions of fibroblast growth factor receptor with its ligands[J]. Proceedings of the National Academy of Sciences of the United States of America. 2000, 97 (1): 49–54
[2] Santos-Ocampo S, Colvin JS, et al. Expression and biological activity of mouse fibroblast growth factor-9[J]. The Journal of Biological Chemistry. 1996, 271 (3): 1726–31
[3] Ornitz DM, Xu J, et al. Receptor specificity of the fibroblast growth factor family [J]. The Journal of Biological Chemistry. 1996, 271(25):15292-15297
[4] Duchesne L, Tissot B, et al. N-glycosylation of fibroblast growth factor receptor 1 regulates ligand and heparan sulfate co-receptor binding [J]. The Journal of Biological Chemistry. 2006, 281(37):27178-27189
[5] Davidson D, Blanc A, et al. Fibroblast growth factor (FGF) 18 signals through FGF receptor 3 to promote chondrogenesis[J]. The Journal of Biological Chemistry. 2005, 280(21):20509-20515
[6] Chellaiah A, Yuan W, et al. Mapping ligand binding domains in chimeric fibroblast growth factor receptor molecules. Multiple regions determine ligand binding specificity[J]. The Journal of Biological Chemistry. 1999, 274 (49): 34785–94
[7] Pavel Krejci, Jirina Prochazkoval, et al. Molecular pathology of the fibroblast growth factor family[J]. Hum Mutat. 2009, 30(9): 1245–1255
[8] Laureys G, Barton DE, et al. Chromosomal mapping of the gene for the type II insulin-like growth factor receptor/cation-independent mannose 6-phosphate receptor in man and mouse[J]. Genomics.1988, 3 (3): 224–9
[9] Ojeda, S. R.; Ma, Y. J., et al. The transforming growth factor alpha gene family is involved in the neuroendocrine control of mammalian puberty[J]. Molecular Psychiatry. 1997, 2 (5): 355–358
[10] Gilchrist, R., Lane, M., et al. Oocyte-secreted factors: regulators of cumulus cell function and oocyte quality[J]. Human Reproduction Update. 2008, 14(2), pp.159-177
[11] Yun, Y.-R., Won, J. E., et al. Fibroblast growth factors: biology, function, and application for tissue regeneration[J]. Tissue Eng. 2010, 218142
[12] Coleman SJ, Grose RP, et al. Fibroblast growth factor family as a potential target in the treatment of hepatocellular carcinoma[J]. J Hepatocell Carcinoma. 2014, 29;1:43-54
[13] Ellman MB, An HS, et al. Biological impact of the fibroblast growth factor family on articular cartilage and intervertebral disc homeostasis[J]. Gene. 2008, 15;420(1):82-9
[14] Olsen SK, Garbi M, et al. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs[J]. J Biol Chem. 2003, 278:34226–34236
[15] Mohammadi M, Dikic I, et al. Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction[J]. Mol Cell Biol. 1996; 16:977–989
[16] Goldfarb M. Fibroblast growth factor homologous factors: evolution, structure, and function[J]. Cytokine Growth Factor Rev. 2005; 16:215–220
[17] Roy NM, Sagerstrom GG. An early Fgf signal required for gene expression in the zebrafish hindbrain primordium[J]. Brain Res Dev Brain Res. 2004; 148(1): 27-42
[18] Katoh Y, Katoh M. Comparative genomics on FGF7, FGF10, FGF22, orthologs, and identification of fgf25[J]. Int J Mol Med. 2005; 16(4): 767-70
[19] Schlessinger J. Cell signaling by receptor tyrosine kinases[J]. Cell. 2000, 103:211–25
[20] Nguyen T, Mège RM. N-Cadherin and Fibroblast Growth Factor Receptors crosstalk in the control of developmental and cancer cell migrations[J]. Eur J Cell Biol. 2016, 95(11):415-426
[21] Feng S, Zhou L, et al. Fibroblast growth factor receptors: multifactorial-contributors to tumor initiation and progression[J]. Histol Histopathol. 2015, 30(1):13-31
[22] Haugsten EM, Wiedlocha A, et al. Roles of fibroblast growth factor receptors in carcinogenesis[J]. Mol Cancer Res. 2010, 8(11):1439-52
[23] Sanak M. Molecular genetics of congenital skeletal dysplasias related to mutations of fibroblast growth factor receptors[J]. Med Wieku Rozwoj. 1999, 3(1):67-82
[24] Wang S, Ding Z. Fibroblast growth factor receptors in breast cancer[J]. Tumour Biol. 2017, 39(5):1010428317698370
[25] Morales-Barrera R, Suárez C, et al. Targeting fibroblast growth factor receptors and immune checkpoint inhibitors for the treatment of advanced bladder cancer: New direction and New Hope[J]. Cancer Treat Rev. 2016, 50:208-216
[26] Inokuchi M, Fujimori Y, et al Therapeutic targeting of fibroblast growth factor receptors in gastric cancer[J]. Gastroenterol Res Pract. 2015, 2015:796380
[27] Sonpavde G, Willey CD, et al. Fibroblast growth factor receptors as therapeutic targets in clear-cell renal cell carcinoma[J]. Expert Opin Investig Drugs. 2014, 23(3):305-15
[28] Kelleher FC, O’Sullivan H, et al. Fibroblast growth factor receptors, developmental corruption and malignant disease[J]. Carcinogenesis. 2013, 34(10):2198-205
[29] Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases[J]. Cell. 2010, 141:1117–1134
[30] Potter JD. Colorectal cancer: molecules and populations[J]. J Natl. Cancer Inst. 1999, 91:916–932
[31] Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression[J]. Nat Rev Cancer 2010, 10:415-424
[32] Huakang Tu, Thomas U. Ahearn, et al. Transforming Growth Factors and Receptor as Potential Modifiable Pre-Neoplastic Biomarkers of Risk for Colorectal Neoplasms[J]. MOLECULAR CARCINOGENESIS. 2015, 54:821-830
[33] Bellone G, Gramigni C, et al. Abnormal expression of Endoglin and its receptor complex (TGF-beta1 and TGF-beta receptor II) as early angiogenic switch indicator in premalignant lesions of the colon mucosa[J]. Int J Oncol. 2010, 37:1153–1165
[34] Hawinkels LJ, Verspaget HW, et al. Active TGF-beta1 correlates with myofibroblasts and malignancy in the colorectal adenoma-carcinoma sequence[J]. Cancer Sci. 2009, 100:663–670
[35] Li Y, Zhu G, et al. Simultaneous stimulation with tumor necrosis factor-α and transforming growth factor-β1 induces epithelial-mesenchymal transition in colon cancer cells via the NF-κB pathway[J]. Oncol Lett. 2018, 15(5):6873-6880
[36] Badawy AA, El-Hindawi A, et al. Impact of epidermal growth factor receptor and transforming growth factor-α on hepatitis C virus-induced hepatocarcinogenesis[J]. APMIS. 2015, 123(10):823-31
[37] Walker F, Abramowitz L, et al. Growth factor receptor expression in anal squamous lesions: modifications associated with oncogenic human papillomavirus and human immunodeficiency virus[J]. Human Pathology. 2009, 40 (11): 1517–27
[38] Midgley AC, Rogers M, et al. Transforming growth factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts[J]. The Journal of Biological Chemistry[J]. 2013, 288 (21): 14824–38
[39] Holmes K, Roberts OL, et al. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition[J]. Cell Signal. 2007, 19 (10): 2003–2012
[40] Zygmunt T, Gay CM, et al. Semaphorin-PlexinD1 Signaling Limits Angiogenic Potential via the VEGF Decoy Receptor sFlt1[J]. Dev Cell. 2011, 21 (2): 301–314
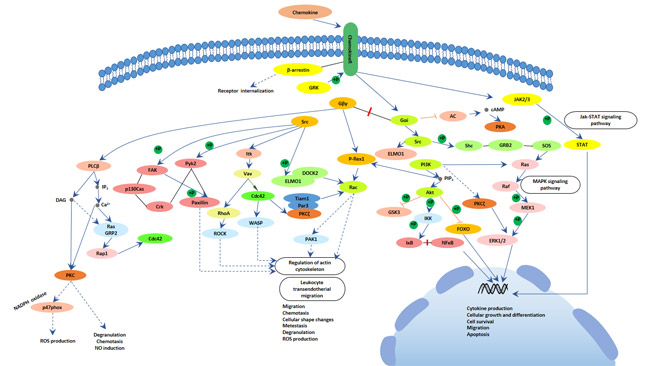
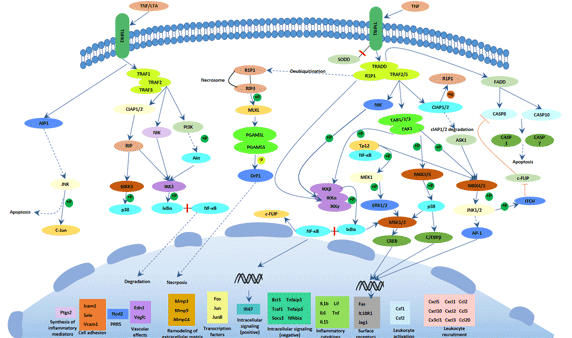
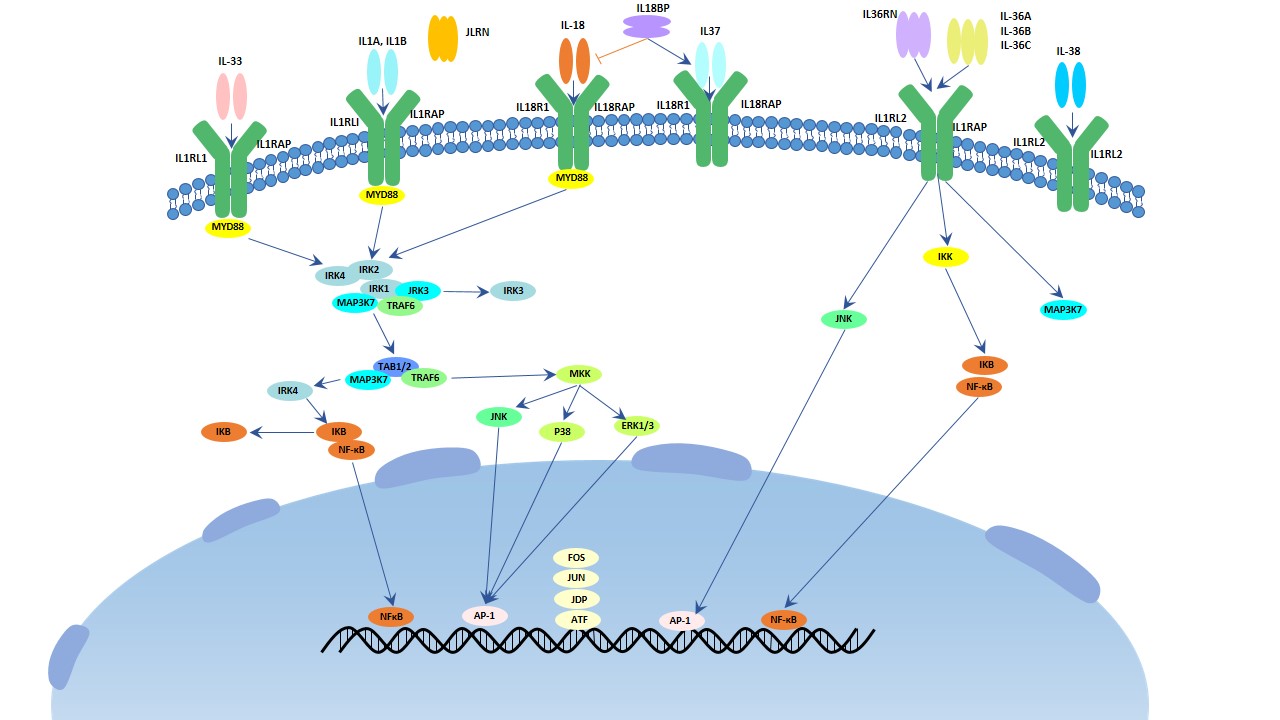
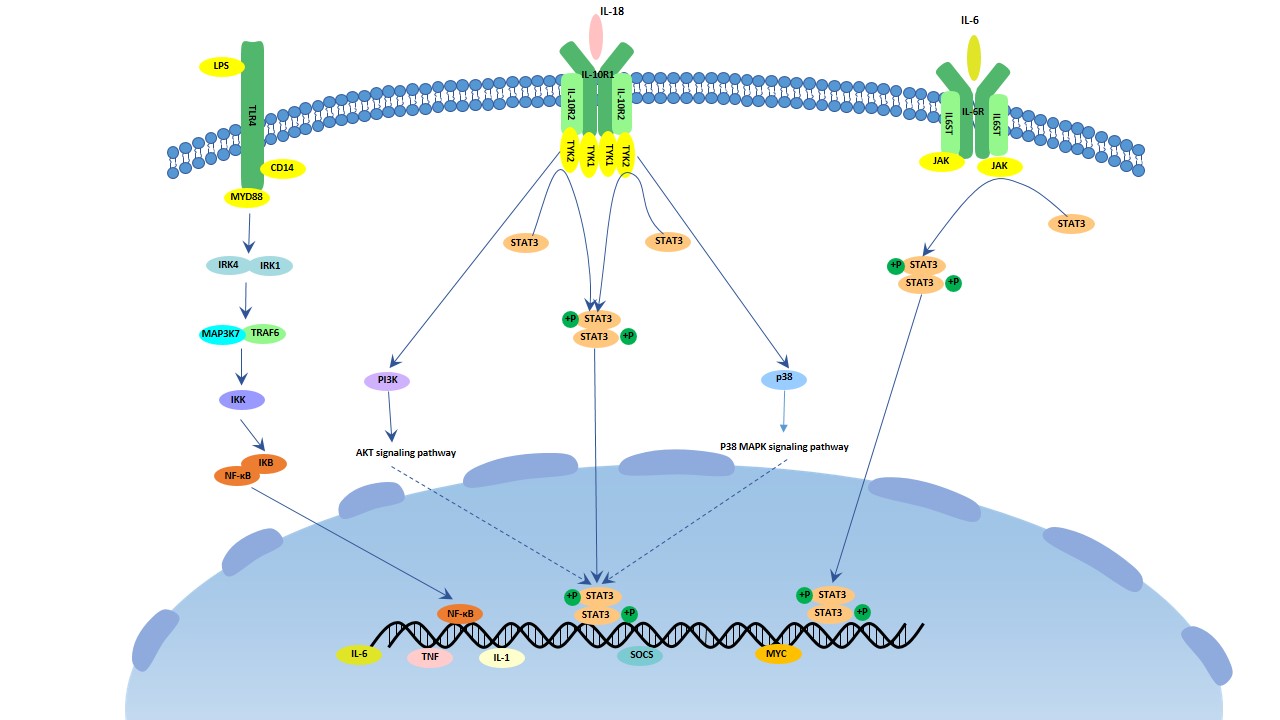
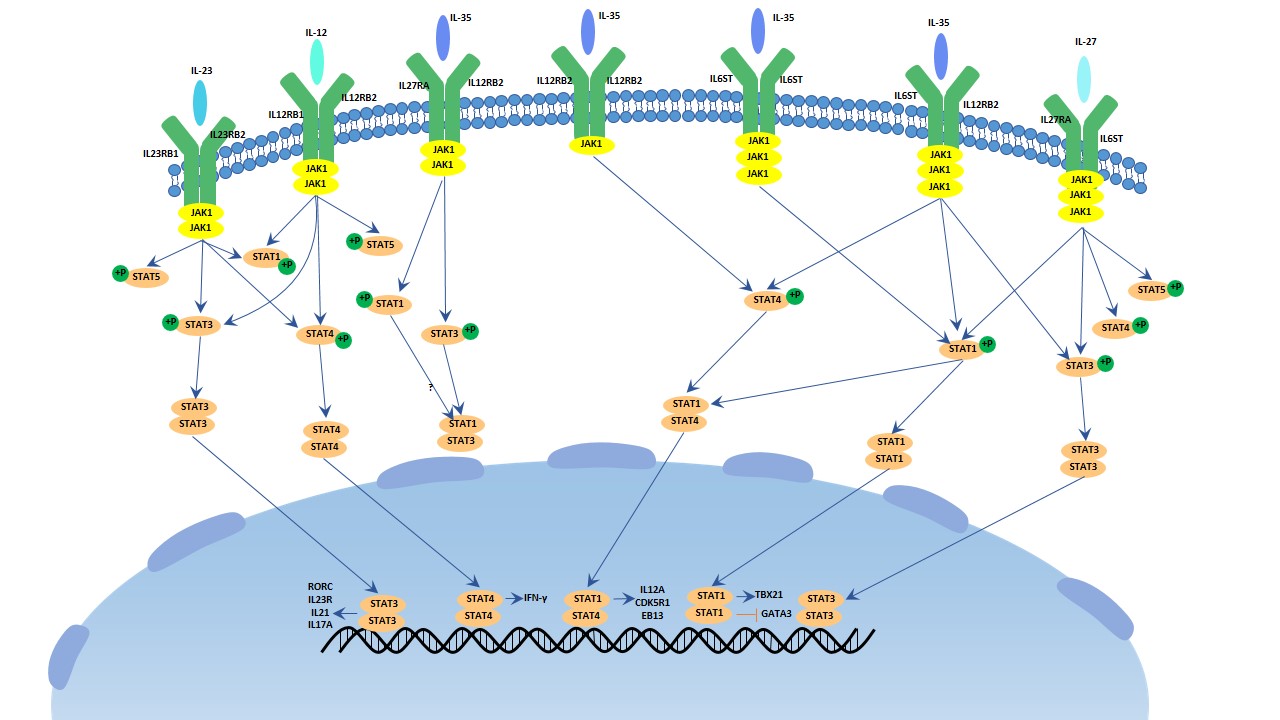
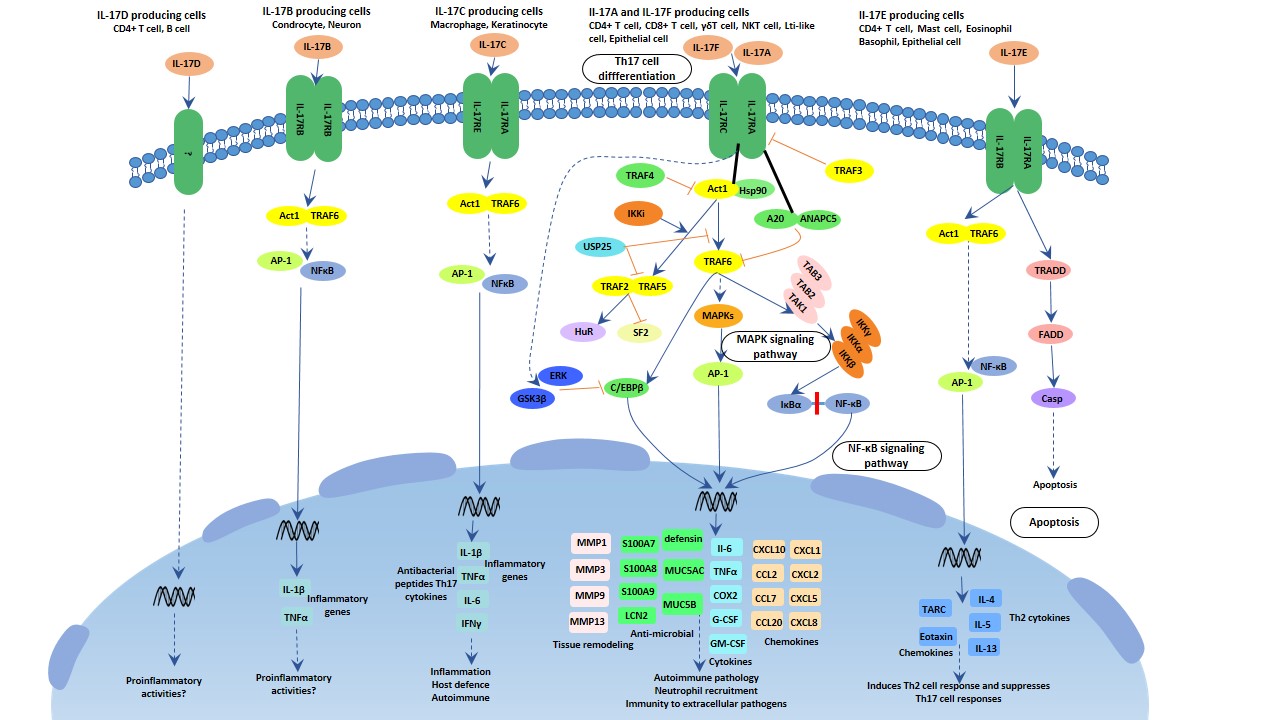
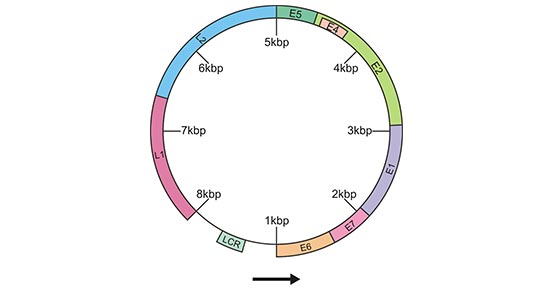
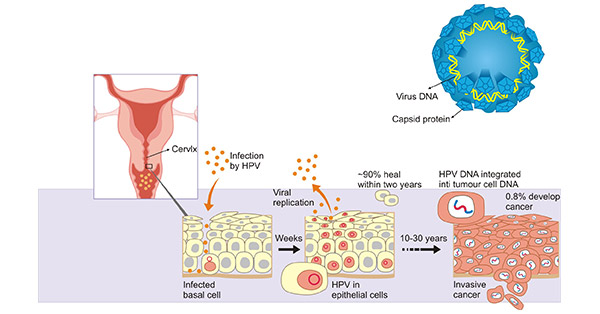
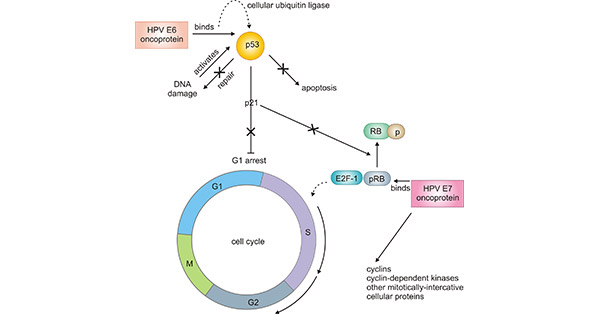
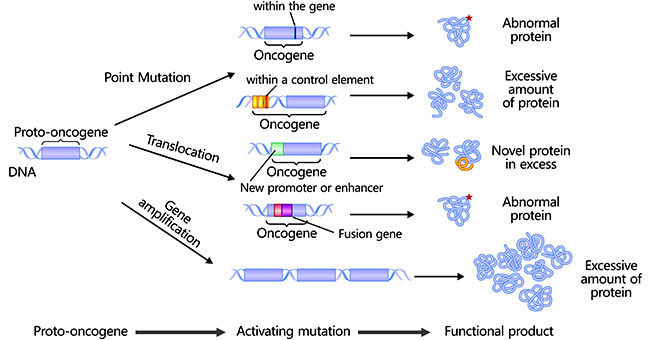
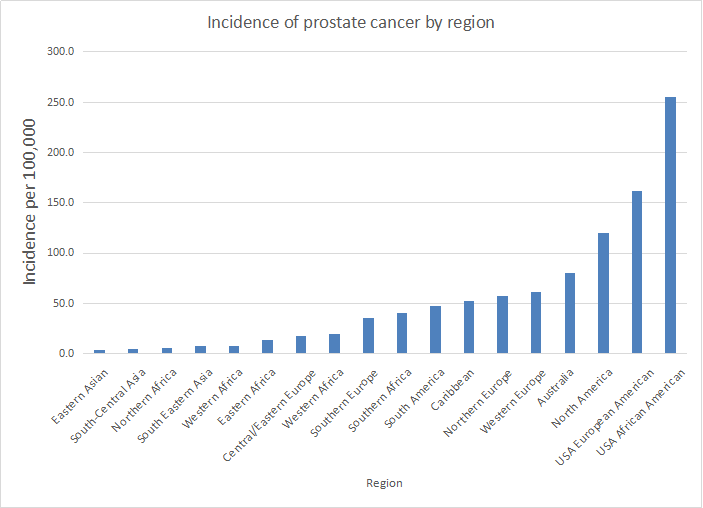
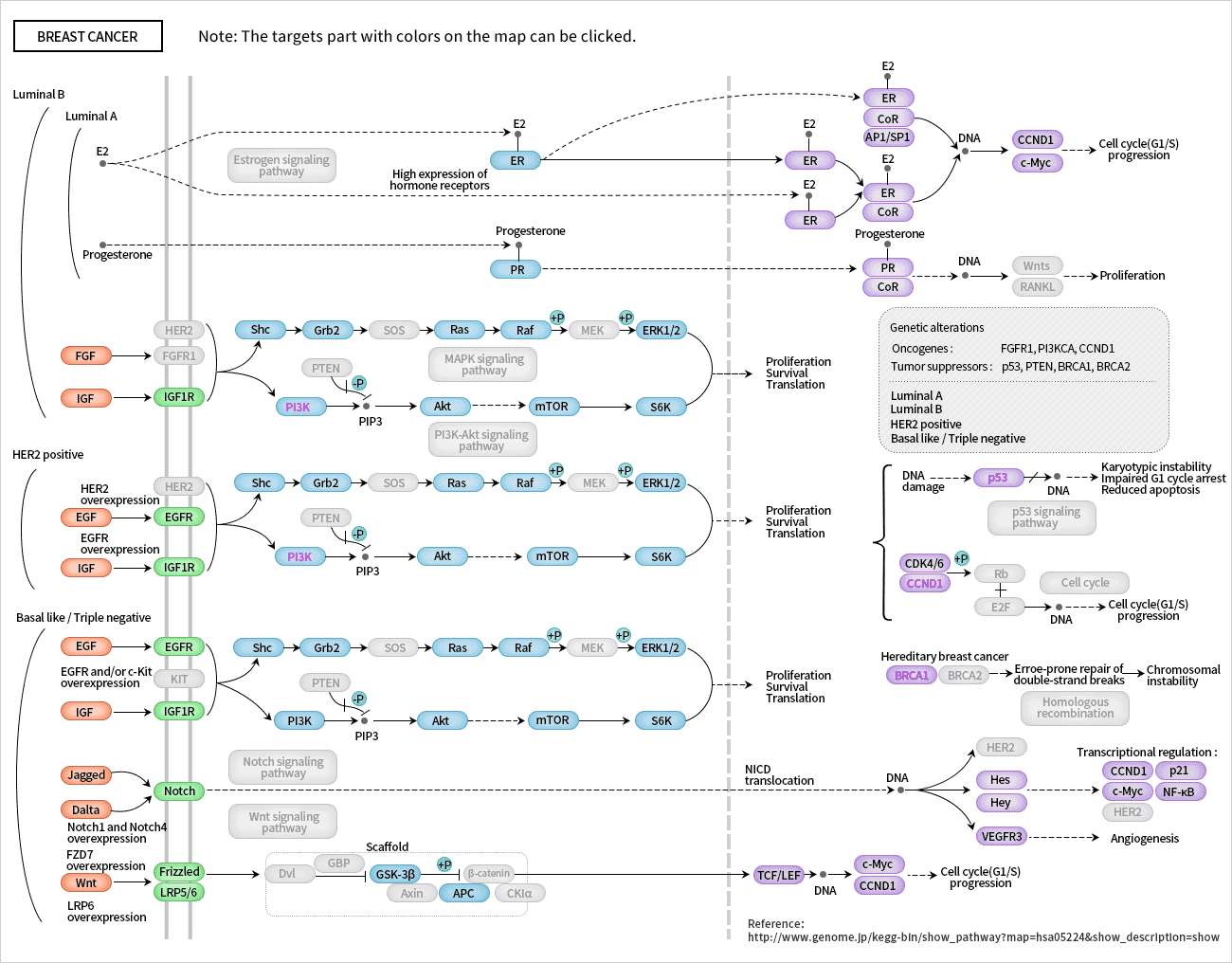

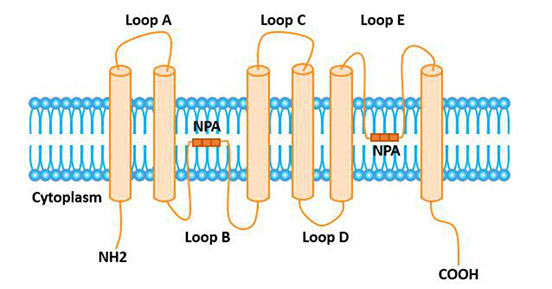
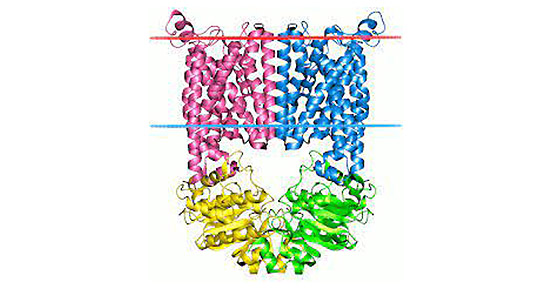


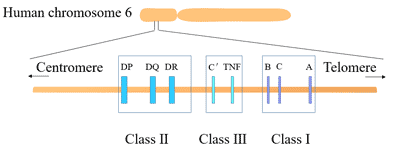
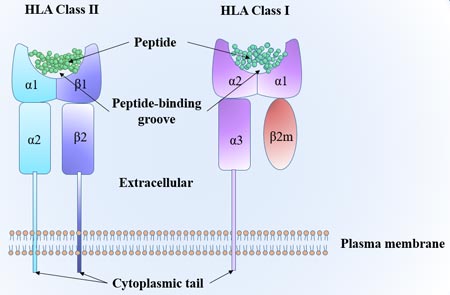
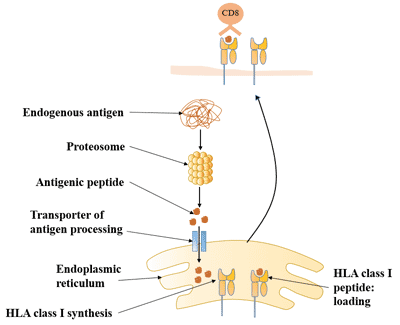
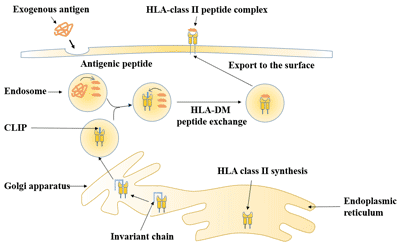

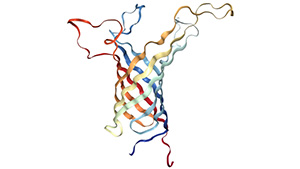
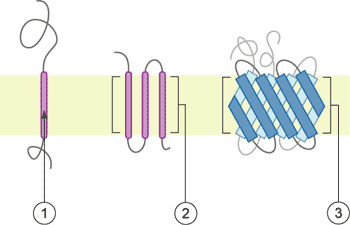
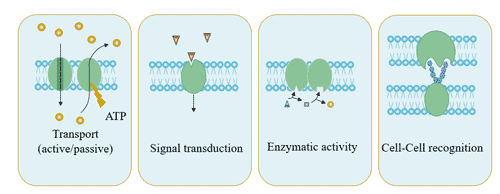

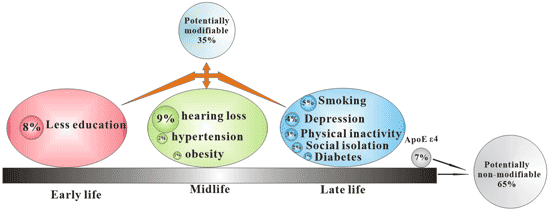
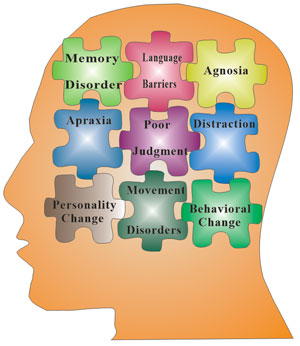

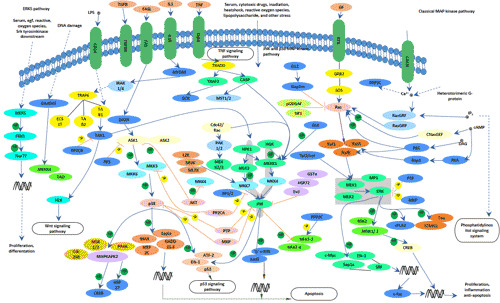
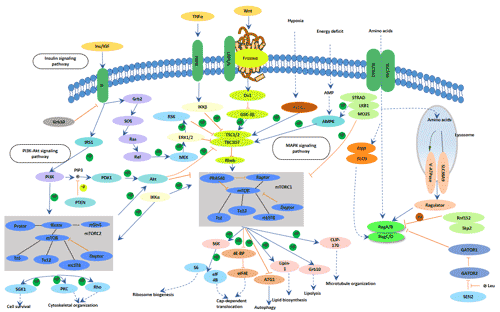
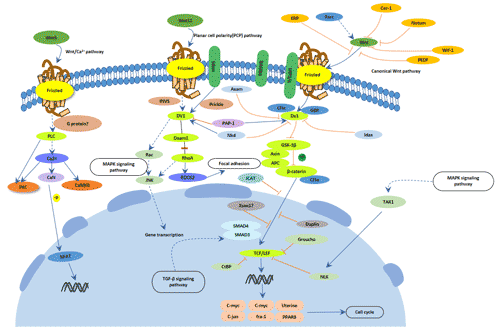
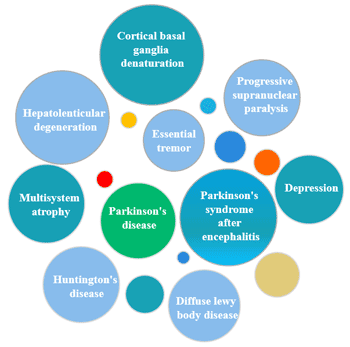 Figure 1 Diseases easily confused with Parkinson’s
Figure 1 Diseases easily confused with Parkinson’s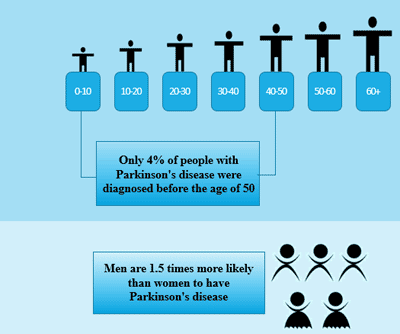
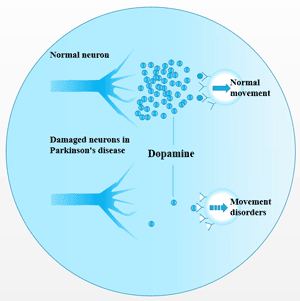
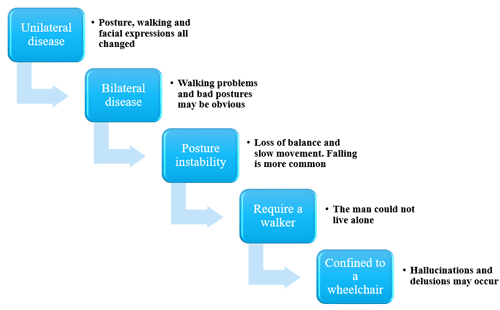
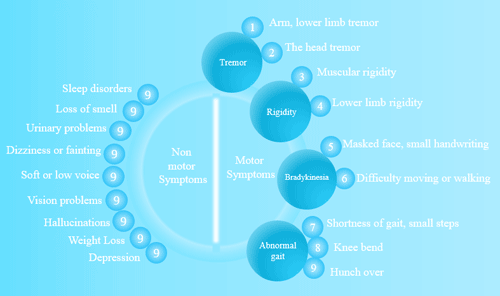
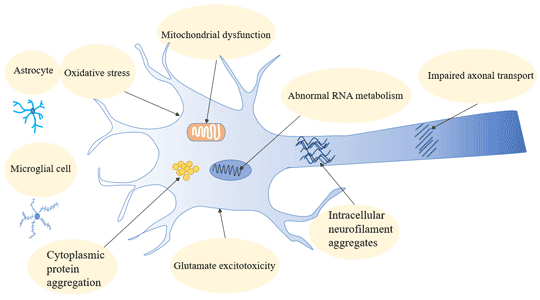
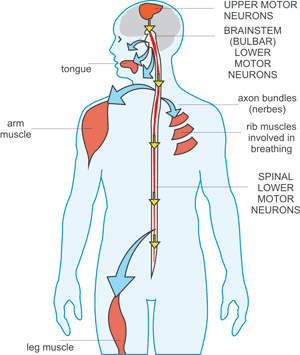
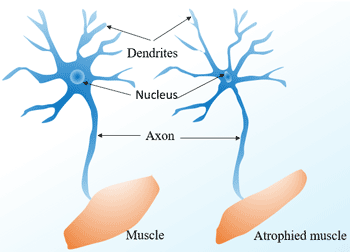
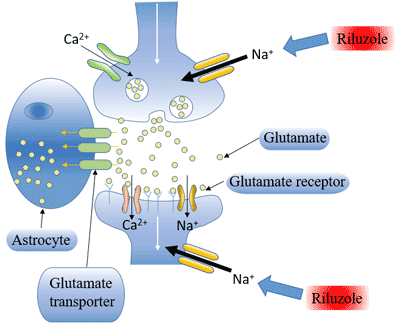
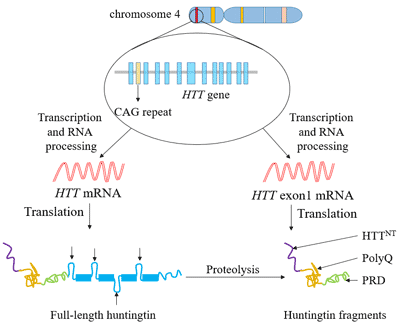
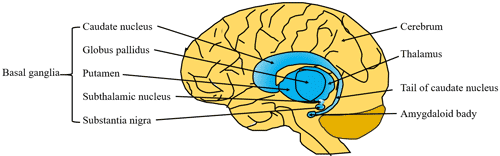
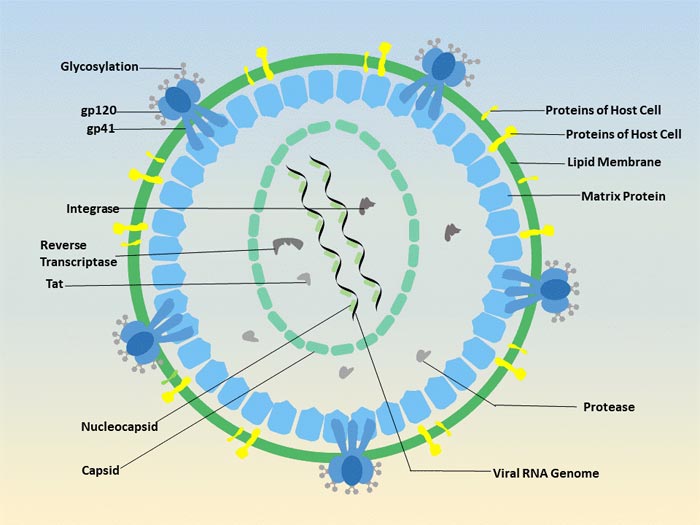
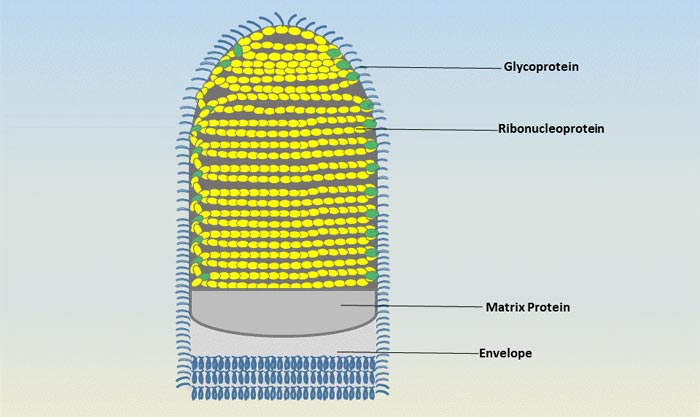
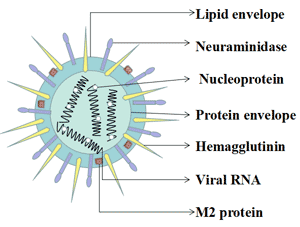
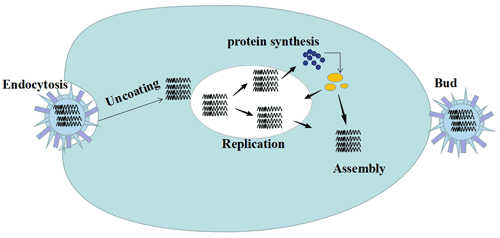
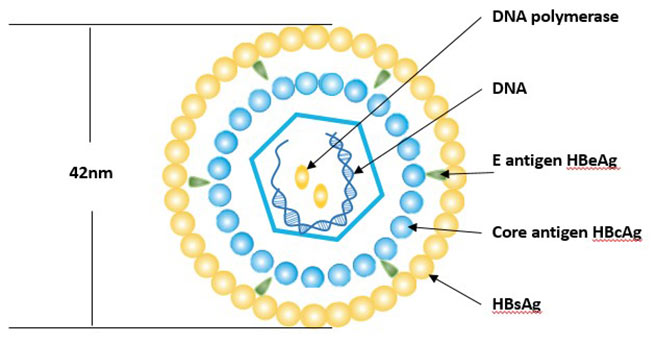
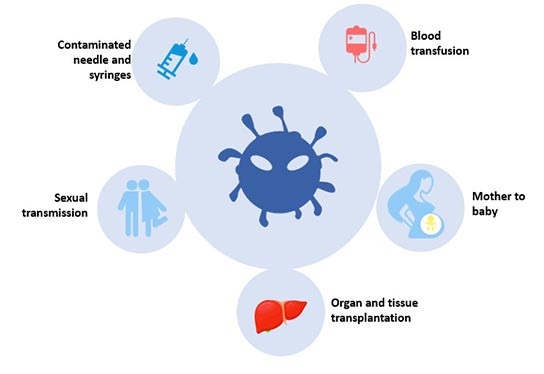 Figure 3 Transmission of hepatitis B infection
Figure 3 Transmission of hepatitis B infection
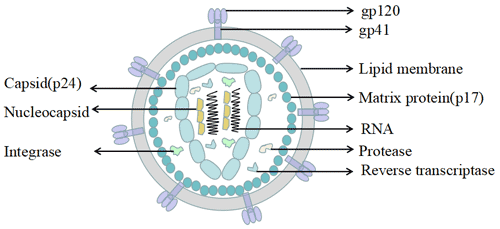
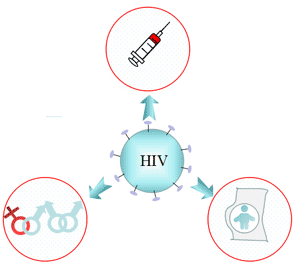
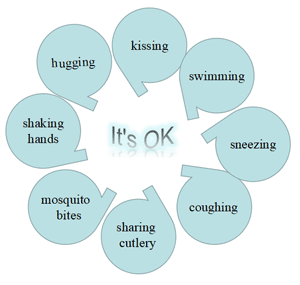
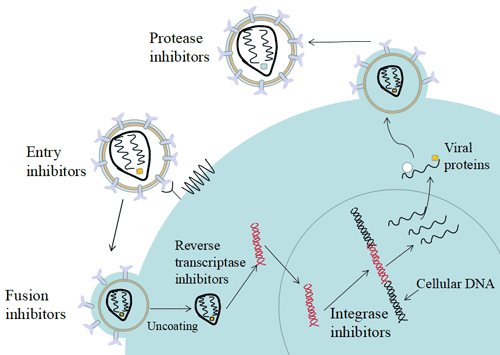
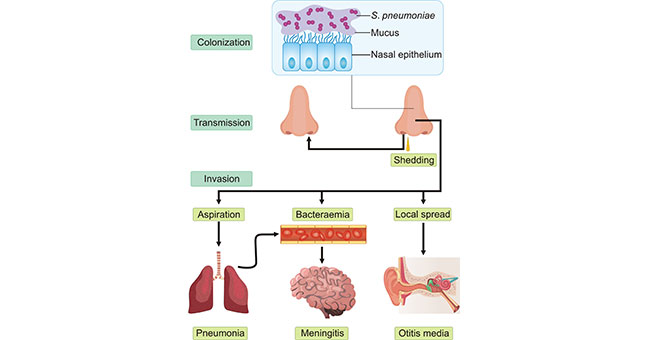
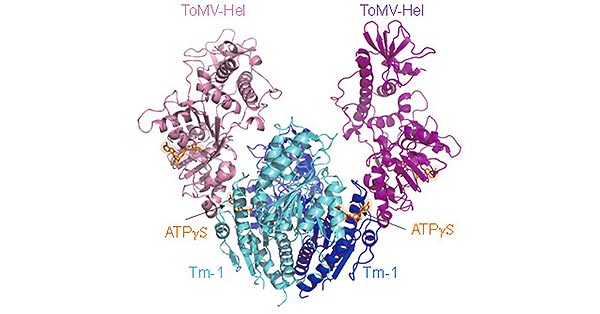
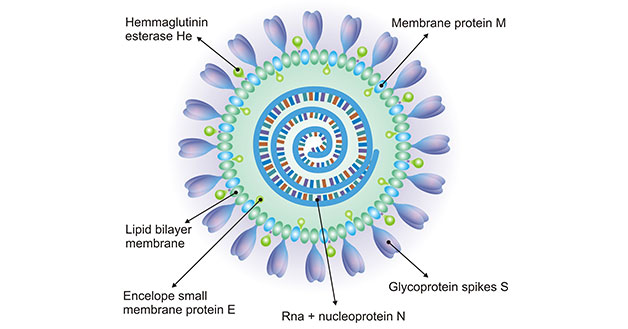
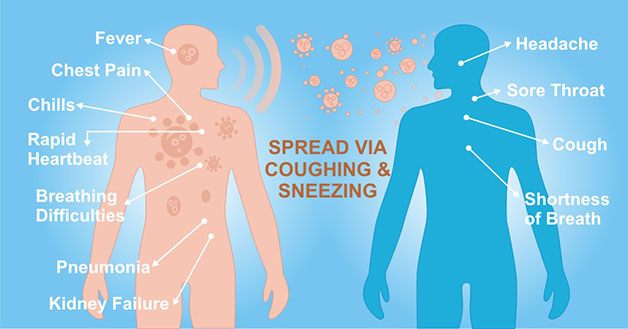
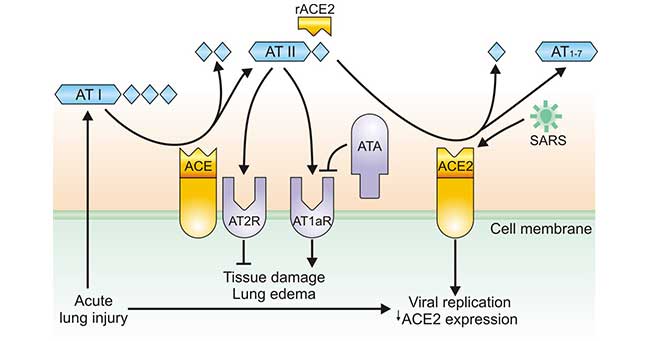
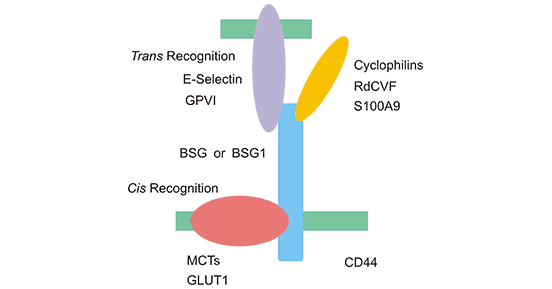
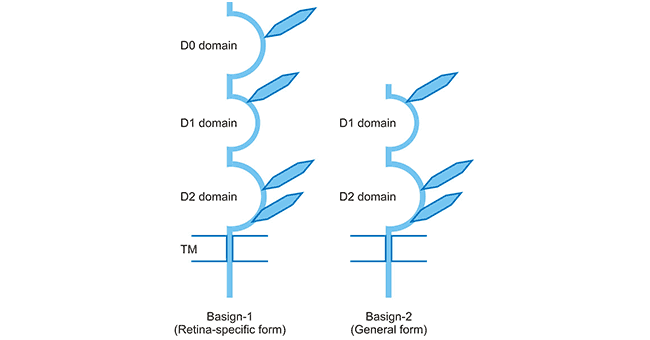
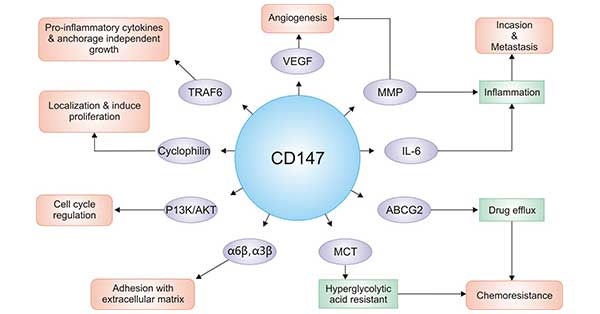
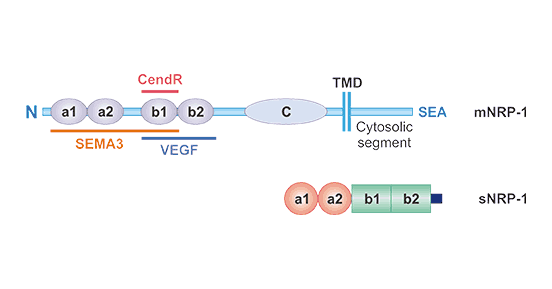 Figure 1. The structure of Neuropilin-1 (NRP1)
Figure 1. The structure of Neuropilin-1 (NRP1)